Use this function to determine a valid microorganism code (mo). Determination is done using intelligent rules and the complete taxonomic kingdoms Animalia, Archaea, Bacteria and Protozoa, and most microbial species from the kingdom Fungi (see Source). The input can be almost anything: a full name (like "Staphylococcus aureus"), an abbreviated name (such as "S. aureus"), an abbreviation known in the field (such as "MRSA"), or just a genus. See Examples.
Usage
as.mo(
x,
Becker = FALSE,
Lancefield = FALSE,
minimum_matching_score = NULL,
keep_synonyms = getOption("AMR_keep_synonyms", FALSE),
reference_df = get_mo_source(),
ignore_pattern = getOption("AMR_ignore_pattern", NULL),
remove_from_input = mo_cleaning_regex(),
language = get_AMR_locale(),
info = interactive(),
...
)
is.mo(x)
mo_uncertainties()
mo_renamed()
mo_failures()
mo_reset_session()
mo_cleaning_regex()Arguments
- x
a character vector or a data.frame with one or two columns
- Becker
a logical to indicate whether staphylococci should be categorised into coagulase-negative staphylococci ("CoNS") and coagulase-positive staphylococci ("CoPS") instead of their own species, according to Karsten Becker et al. (see Source).
This excludes Staphylococcus aureus at default, use
Becker = "all"to also categorise S. aureus as "CoPS".- Lancefield
a logical to indicate whether a beta-haemolytic Streptococcus should be categorised into Lancefield groups instead of their own species, according to Rebecca C. Lancefield (see Source). These streptococci will be categorised in their first group, e.g. Streptococcus dysgalactiae will be group C, although officially it was also categorised into groups G and L.
This excludes enterococci at default (who are in group D), use
Lancefield = "all"to also categorise all enterococci as group D.- minimum_matching_score
a numeric value to set as the lower limit for the MO matching score. When left blank, this will be determined automatically based on the character length of
x, its taxonomic kingdom and human pathogenicity.- keep_synonyms
a logical to indicate if old, previously valid taxonomic names must be preserved and not be corrected to currently accepted names. The default is
FALSE, which will return a note if old taxonomic names were processed. The default can be set withoptions(AMR_keep_synonyms = TRUE)oroptions(AMR_keep_synonyms = FALSE).- reference_df
a data.frame to be used for extra reference when translating
xto a validmo. Seeset_mo_source()andget_mo_source()to automate the usage of your own codes (e.g. used in your analysis or organisation).- ignore_pattern
a regular expression (case-insensitive) of which all matches in
xmust returnNA. This can be convenient to exclude known non-relevant input and can also be set with the optionAMR_ignore_pattern, e.g.options(AMR_ignore_pattern = "(not reported|contaminated flora)").- remove_from_input
a regular expression (case-insensitive) to clean the input of
x. Everything matched inxwill be removed. At default, this is the outcome ofmo_cleaning_regex(), which removes texts between brackets and texts such as "species" and "serovar".- language
language to translate text like "no growth", which defaults to the system language (see
get_AMR_locale())- info
a logical to indicate if a progress bar should be printed if more than 25 items are to be coerced, defaults to
TRUEonly in interactive mode- ...
other arguments passed on to functions
Details
A microorganism (MO) code from this package (class: mo) is human readable and typically looks like these examples:
Code Full name
--------------- --------------------------------------
B_KLBSL Klebsiella
B_KLBSL_PNMN Klebsiella pneumoniae
B_KLBSL_PNMN_RHNS Klebsiella pneumoniae rhinoscleromatis
| | | |
| | | |
| | | \---> subspecies, a 3-5 letter acronym
| | \----> species, a 3-6 letter acronym
| \----> genus, a 4-8 letter acronym
\----> taxonomic kingdom: A (Archaea), AN (Animalia), B (Bacteria),
F (Fungi), PL (Plantae), P (Protozoa)Values that cannot be coerced will be considered 'unknown' and will be returned as the MO code UNKNOWN with a warning.
Use the mo_* functions to get properties based on the returned code, see Examples.
The as.mo() function uses a novel matching score algorithm (see Matching Score for Microorganisms below) to match input against the available microbial taxonomy in this package. This will lead to the effect that e.g. "E. coli" (a microorganism highly prevalent in humans) will return the microbial ID of Escherichia coli and not Entamoeba coli (a microorganism less prevalent in humans), although the latter would alphabetically come first. The algorithm uses data from the List of Prokaryotic names with Standing in Nomenclature (LPSN) and the Global Biodiversity Information Facility (GBIF) (see microorganisms).
Coping with Uncertain Results
Results of non-exact taxonomic input are based on their matching score. The lowest allowed score can be set with the minimum_matching_score argument. At default this will be determined based on the character length of the input, and the taxonomic kingdom and human pathogenicity of the taxonomic outcome. If values are matched with uncertainty, a message will be shown to suggest the user to evaluate the results with mo_uncertainties(), which returns a data.frame with all specifications.
To increase the quality of matching, the remove_from_input argument can be used to clean the input (i.e., x). This must be a regular expression that matches parts of the input that should be removed before the input is matched against the available microbial taxonomy. It will be matched Perl-compatible and case-insensitive. The default value of remove_from_input is the outcome of the helper function mo_cleaning_regex().
There are three helper functions that can be run after using the as.mo() function:
Use
mo_uncertainties()to get a data.frame that prints in a pretty format with all taxonomic names that were guessed. The output contains the matching score for all matches (see Matching Score for Microorganisms below).Use
mo_failures()to get a character vector with all values that could not be coerced to a valid value.Use
mo_renamed()to get a data.frame with all values that could be coerced based on old, previously accepted taxonomic names.
Microbial Prevalence of Pathogens in Humans
The coercion rules consider the prevalence of microorganisms in humans grouped into three groups, which is available as the prevalence columns in the microorganisms data set. The grouping into human pathogenic prevalence is explained in the section Matching Score for Microorganisms below.
Source
Berends MS et al. (2022). AMR: An R Package for Working with Antimicrobial Resistance Data. Journal of Statistical Software, 104(3), 1-31; doi:10.18637/jss.v104.i03
Becker K et al. (2014). Coagulase-Negative Staphylococci. Clin Microbiol Rev. 27(4): 870-926; doi:10.1128/CMR.00109-13
Becker K et al. (2019). Implications of identifying the recently defined members of the S. aureus complex, S. argenteus and S. schweitzeri: A position paper of members of the ESCMID Study Group for staphylococci and Staphylococcal Diseases (ESGS). Clin Microbiol Infect; doi:10.1016/j.cmi.2019.02.028
Becker K et al. (2020). Emergence of coagulase-negative staphylococci Expert Rev Anti Infect Ther. 18(4):349-366; doi:10.1080/14787210.2020.1730813
Lancefield RC (1933). A serological differentiation of human and other groups of hemolytic streptococci. J Exp Med. 57(4): 571-95; doi:10.1084/jem.57.4.571
Berends MS et al. (2022). Trends in Occurrence and Phenotypic Resistance of Coagulase-Negative Staphylococci (CoNS) Found in Human Blood in the Northern Netherlands between 2013 and 2019 Microorganisms 10(9), 1801; doi:10.3390/microorganisms10091801
Parte, AC et al. (2020). List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. International Journal of Systematic and Evolutionary Microbiology, 70, 5607-5612; doi:10.1099/ijsem.0.004332 . Accessed from https://lpsn.dsmz.de on 12 September, 2022.
GBIF Secretariat (November 26, 2021). GBIF Backbone Taxonomy. Checklist dataset doi:10.15468/39omei . Accessed from https://www.gbif.org on 12 September, 2022.
Public Health Information Network Vocabulary Access and Distribution System (PHIN VADS). US Edition of SNOMED CT from 1 September 2020. Value Set Name 'Microoganism', OID 2.16.840.1.114222.4.11.1009 (v12). URL: https://phinvads.cdc.gov
Matching Score for Microorganisms
With ambiguous user input in as.mo() and all the mo_* functions, the returned results are chosen based on their matching score using mo_matching_score(). This matching score \(m\), is calculated as:
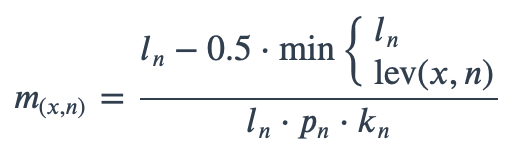
where:
x is the user input;
n is a taxonomic name (genus, species, and subspecies);
ln is the length of n;
lev is the Levenshtein distance function (counting any insertion as 1, and any deletion or substitution as 2) that is needed to change x into n;
pn is the human pathogenic prevalence group of n, as described below;
kn is the taxonomic kingdom of n, set as Bacteria = 1, Fungi = 2, Protozoa = 3, Archaea = 4, others = 5.
The grouping into human pathogenic prevalence (\(p\)) is based on experience from several microbiological laboratories in the Netherlands in conjunction with international reports on pathogen prevalence:
Group 1 (most prevalent microorganisms) consists of all microorganisms where the taxonomic class is Gammaproteobacteria or where the taxonomic genus is Enterococcus, Staphylococcus or Streptococcus. This group consequently contains all common Gram-negative bacteria, such as Pseudomonas and Legionella and all species within the order Enterobacterales.
Group 2 consists of all microorganisms where the taxonomic phylum is Proteobacteria, Firmicutes, Actinobacteria or Sarcomastigophora, or where the taxonomic genus is Absidia, Acanthamoeba, Acholeplasma, Acremonium, Actinotignum, Aedes, Alistipes, Alloprevotella, Alternaria, Amoeba, Anaerosalibacter, Ancylostoma, Angiostrongylus, Anisakis, Anopheles, Apophysomyces, Arachnia, Aspergillus, Aureobasidium, Bacteroides, Basidiobolus, Beauveria, Bergeyella, Blastocystis, Blastomyces, Borrelia, Brachyspira, Branhamella, Butyricimonas, Candida, Capillaria, Capnocytophaga, Catabacter, Cetobacterium, Chaetomium, Chlamydia, Chlamydophila, Chryseobacterium, Chrysonilia, Cladophialophora, Cladosporium, Conidiobolus, Contracaecum, Cordylobia, Cryptococcus, Curvularia, Deinococcus, Demodex, Dermatobia, Dientamoeba, Diphyllobothrium, Dirofilaria, Dysgonomonas, Echinostoma, Elizabethkingia, Empedobacter, Entamoeba, Enterobius, Exophiala, Exserohilum, Fasciola, Flavobacterium, Fonsecaea, Fusarium, Fusobacterium, Giardia, Haloarcula, Halobacterium, Halococcus, Hendersonula, Heterophyes, Histomonas, Histoplasma, Hymenolepis, Hypomyces, Hysterothylacium, Leishmania, Lelliottia, Leptosphaeria, Leptotrichia, Lucilia, Lumbricus, Malassezia, Malbranchea, Metagonimus, Meyerozyma, Microsporidium, Microsporum, Mortierella, Mucor, Mycocentrospora, Mycoplasma, Myroides, Necator, Nectria, Ochroconis, Odoribacter, Oesophagostomum, Oidiodendron, Opisthorchis, Ornithobacterium, Parabacteroides, Pediculus, Pedobacter, Phlebotomus, Phocaeicola, Phocanema, Phoma, Pichia, Piedraia, Pithomyces, Pityrosporum, Pneumocystis, Porphyromonas, Prevotella, Pseudallescheria, Pseudoterranova, Pulex, Rhizomucor, Rhizopus, Rhodotorula, Riemerella, Saccharomyces, Sarcoptes, Scolecobasidium, Scopulariopsis, Scytalidium, Sphingobacterium, Spirometra, Spiroplasma, Sporobolomyces, Stachybotrys, Streptobacillus, Strongyloides, Syngamus, Taenia, Tannerella, Tenacibaculum, Terrimonas, Toxocara, Treponema, Trichinella, Trichobilharzia, Trichoderma, Trichomonas, Trichophyton, Trichosporon, Trichostrongylus, Trichuris, Tritirachium, Trombicula, Trypanosoma, Tunga, Ureaplasma, Victivallis, Wautersiella, Weeksella or Wuchereria.
Group 3 consists of all other microorganisms.
All characters in \(x\) and \(n\) are ignored that are other than A-Z, a-z, 0-9, spaces and parentheses.
All matches are sorted descending on their matching score and for all user input values, the top match will be returned. This will lead to the effect that e.g., "E. coli" will return the microbial ID of Escherichia coli (\(m = 0.688\), a highly prevalent microorganism found in humans) and not Entamoeba coli (\(m = 0.119\), a less prevalent microorganism in humans), although the latter would alphabetically come first.
Reference Data Publicly Available
All data sets in this AMR package (about microorganisms, antibiotics, R/SI interpretation, EUCAST rules, etc.) are publicly and freely available for download in the following formats: R, MS Excel, Apache Feather, Apache Parquet, SPSS, SAS, and Stata. We also provide tab-separated plain text files that are machine-readable and suitable for input in any software program, such as laboratory information systems. Please visit our website for the download links. The actual files are of course available on our GitHub repository.
See also
microorganisms for the data.frame that is being used to determine ID's.
The mo_* functions (such as mo_genus(), mo_gramstain()) to get properties based on the returned code.
Examples
# \donttest{
# These examples all return "B_STPHY_AURS", the ID of S. aureus:
as.mo(c(
"sau", # WHONET code
"stau",
"STAU",
"staaur",
"S. aureus",
"S aureus",
"Sthafilokkockus aureus", # handles incorrect spelling
"Staphylococcus aureus (MRSA)",
"MRSA", # Methicillin Resistant S. aureus
"VISA", # Vancomycin Intermediate S. aureus
"VRSA", # Vancomycin Resistant S. aureus
115329001 # SNOMED CT code
))
#> Class <mo>
#> [1] B_STPHY_AURS B_STPHY_AURS B_STPHY_AURS B_STPHY_AURS B_STPHY_AURS
#> [6] B_STPHY_AURS B_STPHY_AURS B_STPHY_AURS B_STPHY_AURS B_STPHY_AURS
#> [11] B_STPHY_AURS B_STPHY_AURS
# Dyslexia is no problem - these all work:
as.mo(c(
"Ureaplasma urealyticum",
"Ureaplasma urealyticus",
"Ureaplasmium urealytica",
"Ureaplazma urealitycium"
))
#> Class <mo>
#> [1] B_URPLS_URLY B_URPLS_URLY B_URPLS_URLY B_URPLS_URLY
as.mo("Streptococcus group A")
#> Class <mo>
#> [1] B_STRPT_GRPA
as.mo("S. epidermidis") # will remain species: B_STPHY_EPDR
#> Class <mo>
#> [1] B_STPHY_EPDR
as.mo("S. epidermidis", Becker = TRUE) # will not remain species: B_STPHY_CONS
#> Class <mo>
#> [1] B_STPHY_CONS
as.mo("S. pyogenes") # will remain species: B_STRPT_PYGN
#> Class <mo>
#> [1] B_STRPT_PYGN
as.mo("S. pyogenes", Lancefield = TRUE) # will not remain species: B_STRPT_GRPA
#> Class <mo>
#> [1] B_STRPT_GRPA
# All mo_* functions use as.mo() internally too (see ?mo_property):
mo_genus("E. coli")
#> [1] "Escherichia"
mo_gramstain("ESCO")
#> [1] "Gram-negative"
mo_is_intrinsic_resistant("ESCCOL", ab = "vanco")
#> ℹ Determining intrinsic resistance based on 'EUCAST Expert Rules' and
#> 'EUCAST Intrinsic Resistance and Unusual Phenotypes' v3.3 (2021). This note
#> will be shown once per session.
#> [1] TRUE
# }