# `AMR`
### An [R package](https://www.r-project.org) to simplify the analysis and prediction of Antimicrobial Resistance (AMR) and work with antibiotic properties by using evidence-based methods.
This R package was created for academic research by PhD students of the Faculty of Medical Sciences of the [University of Groningen](https://www.rug.nl) and the Medical Microbiology & Infection Prevention (MMBI) department of the [University Medical Center Groningen (UMCG)](https://www.umcg.nl).
:arrow_forward: Download it with `install.packages("AMR")` or see below for other possibilities.
## Authors
 Matthijs S. Berends1,2,a,
Christian F. Luz1,a,
Erwin E.A. Hassing2,
Corinna Glasner1,b,
Alex W. Friedrich1,b,
Bhanu Sinha1,b
1 Department of Medical Microbiology, University of Groningen, University Medical Center Groningen, Groningen, the Netherlands
Matthijs S. Berends1,2,a,
Christian F. Luz1,a,
Erwin E.A. Hassing2,
Corinna Glasner1,b,
Alex W. Friedrich1,b,
Bhanu Sinha1,b
1 Department of Medical Microbiology, University of Groningen, University Medical Center Groningen, Groningen, the Netherlands
2 Certe Medical Diagnostics & Advice, Groningen, the Netherlands
a R package author and dissertant
b Thesis advisor




 ## Why this package?
This R package contains functions to make **microbiological, epidemiological data analysis easier**. It allows the use of some new classes to work with MIC values and antimicrobial interpretations (i.e. values S, I and R).
With `AMR` you can:
* Calculate the resistance (and even co-resistance) of microbial isolates with the `rsi_R`, `rsi_IR`, `rsi_I`, `rsi_SI` and `rsi_S` functions, that can also be used with the `dplyr` package (e.g. in conjunction with `summarise`)
* Predict antimicrobial resistance for the nextcoming years with the `resistance_predict` function
* Apply [EUCAST rules to isolates](http://www.eucast.org/expert_rules_and_intrinsic_resistance/) with the `EUCAST_rules` function
* Identify first isolates of every patient [using guidelines from the CLSI](https://clsi.org/standards/products/microbiology/documents/m39/) (Clinical and Laboratory Standards Institute) with the `first_isolate` function
* You can also identify first *weighted* isolates of every patient, an adjusted version of the CLSI guideline. This takes into account key antibiotics of every strain and compares them. The following 12 antibiotics will be used as key antibiotics at default:
* Universal: amoxicillin, amoxicillin/clavlanic acid, cefuroxime, piperacillin/tazobactam, ciprofloxacin, trimethoprim/sulfamethoxazole
* Specific for Gram-positives: vancomycin, teicoplanin, tetracycline, erythromycin, oxacillin, rifampicin
* Specific for Gram-negatives: gentamicin, tobramycin, colistin, cefotaxime, ceftazidime, meropenem
* Categorise *Staphylococci* into Coagulase Negative *Staphylococci* (CoNS) and Coagulase Positve *Staphylococci* (CoPS) according to [Karsten Becker *et al.*](https://www.ncbi.nlm.nih.gov/pmc/articles/pmid/25278577/)
* Categorise *Streptococci* into Lancefield groups
* Get antimicrobial ATC properties from the WHO Collaborating Centre for Drug Statistics Methodology ([WHOCC](https://www.whocc.no/atc_ddd_methodology/who_collaborating_centre/)), to be able to:
* Translate antibiotic codes (like *AMOX*), official names (like *amoxicillin*) and even trade names (like *Amoxil* or *Trimox*) to an [ATC code](https://www.whocc.no/atc_ddd_index/?code=J01CA04&showdescription=no) (like *J01CA04*) and vice versa with the `abname` function
* Get the latest antibiotic properties like hierarchic groups and [defined daily dose](https://en.wikipedia.org/wiki/Defined_daily_dose) (DDD) with units and administration form from the WHOCC website with the `atc_property` function
* Conduct descriptive statistics: calculate kurtosis, skewness and create frequency tables
And it contains:
* A recent data set with ~2500 human pathogenic microorganisms, including family, genus, species, gram stain and aerobic/anaerobic
* A recent data set with all antibiotics as defined by the [WHOCC](https://www.whocc.no/atc_ddd_methodology/who_collaborating_centre/), including ATC code, official name and DDD's
* An example data set `septic_patients`, consisting of 2000 blood culture isolates from anonymised septic patients between 2001 and 2017.
With the `MDRO` function (abbreviation of Multi Drug Resistant Organisms), you can check your isolates for exceptional resistance with country-specific guidelines or EUCAST rules. Currently guidelines for Germany and the Netherlands are supported. Please suggest addition of your own country here: [https://github.com/msberends/AMR/issues/new](https://github.com/msberends/AMR/issues/new?title=New%20guideline%20for%20MDRO&body=%3C--%20Please%20add%20your%20country%20code,%20guideline%20name,%20version%20and%20source%20below%20and%20remove%20this%20line--%3E).
The functions to calculate microbial resistance use expressions that are not evaluated by R itself, but by alternative C++ code that is 25 to 30 times faster and uses less memory. This is called *hybrid evaluation*.
#### Read all changes and new functions in [NEWS.md](NEWS.md).
## How to get it?
This package [is published on CRAN](http://cran.r-project.org/package=AMR), the official R network.
### Install from CRAN (recommended)
[](http://cran.r-project.org/package=AMR) [](http://cran.r-project.org/package=AMR)
(Note: Downloads measured only by [cran.rstudio.com](https://cran.rstudio.com/package=AMR), this excludes e.g. the official [cran.r-project.org](https://cran.r-project.org/package=AMR))
-
## Why this package?
This R package contains functions to make **microbiological, epidemiological data analysis easier**. It allows the use of some new classes to work with MIC values and antimicrobial interpretations (i.e. values S, I and R).
With `AMR` you can:
* Calculate the resistance (and even co-resistance) of microbial isolates with the `rsi_R`, `rsi_IR`, `rsi_I`, `rsi_SI` and `rsi_S` functions, that can also be used with the `dplyr` package (e.g. in conjunction with `summarise`)
* Predict antimicrobial resistance for the nextcoming years with the `resistance_predict` function
* Apply [EUCAST rules to isolates](http://www.eucast.org/expert_rules_and_intrinsic_resistance/) with the `EUCAST_rules` function
* Identify first isolates of every patient [using guidelines from the CLSI](https://clsi.org/standards/products/microbiology/documents/m39/) (Clinical and Laboratory Standards Institute) with the `first_isolate` function
* You can also identify first *weighted* isolates of every patient, an adjusted version of the CLSI guideline. This takes into account key antibiotics of every strain and compares them. The following 12 antibiotics will be used as key antibiotics at default:
* Universal: amoxicillin, amoxicillin/clavlanic acid, cefuroxime, piperacillin/tazobactam, ciprofloxacin, trimethoprim/sulfamethoxazole
* Specific for Gram-positives: vancomycin, teicoplanin, tetracycline, erythromycin, oxacillin, rifampicin
* Specific for Gram-negatives: gentamicin, tobramycin, colistin, cefotaxime, ceftazidime, meropenem
* Categorise *Staphylococci* into Coagulase Negative *Staphylococci* (CoNS) and Coagulase Positve *Staphylococci* (CoPS) according to [Karsten Becker *et al.*](https://www.ncbi.nlm.nih.gov/pmc/articles/pmid/25278577/)
* Categorise *Streptococci* into Lancefield groups
* Get antimicrobial ATC properties from the WHO Collaborating Centre for Drug Statistics Methodology ([WHOCC](https://www.whocc.no/atc_ddd_methodology/who_collaborating_centre/)), to be able to:
* Translate antibiotic codes (like *AMOX*), official names (like *amoxicillin*) and even trade names (like *Amoxil* or *Trimox*) to an [ATC code](https://www.whocc.no/atc_ddd_index/?code=J01CA04&showdescription=no) (like *J01CA04*) and vice versa with the `abname` function
* Get the latest antibiotic properties like hierarchic groups and [defined daily dose](https://en.wikipedia.org/wiki/Defined_daily_dose) (DDD) with units and administration form from the WHOCC website with the `atc_property` function
* Conduct descriptive statistics: calculate kurtosis, skewness and create frequency tables
And it contains:
* A recent data set with ~2500 human pathogenic microorganisms, including family, genus, species, gram stain and aerobic/anaerobic
* A recent data set with all antibiotics as defined by the [WHOCC](https://www.whocc.no/atc_ddd_methodology/who_collaborating_centre/), including ATC code, official name and DDD's
* An example data set `septic_patients`, consisting of 2000 blood culture isolates from anonymised septic patients between 2001 and 2017.
With the `MDRO` function (abbreviation of Multi Drug Resistant Organisms), you can check your isolates for exceptional resistance with country-specific guidelines or EUCAST rules. Currently guidelines for Germany and the Netherlands are supported. Please suggest addition of your own country here: [https://github.com/msberends/AMR/issues/new](https://github.com/msberends/AMR/issues/new?title=New%20guideline%20for%20MDRO&body=%3C--%20Please%20add%20your%20country%20code,%20guideline%20name,%20version%20and%20source%20below%20and%20remove%20this%20line--%3E).
The functions to calculate microbial resistance use expressions that are not evaluated by R itself, but by alternative C++ code that is 25 to 30 times faster and uses less memory. This is called *hybrid evaluation*.
#### Read all changes and new functions in [NEWS.md](NEWS.md).
## How to get it?
This package [is published on CRAN](http://cran.r-project.org/package=AMR), the official R network.
### Install from CRAN (recommended)
[](http://cran.r-project.org/package=AMR) [](http://cran.r-project.org/package=AMR)
(Note: Downloads measured only by [cran.rstudio.com](https://cran.rstudio.com/package=AMR), this excludes e.g. the official [cran.r-project.org](https://cran.r-project.org/package=AMR))
-  Install using [RStudio](http://www.rstudio.com) (recommended):
- Click on `Tools` and then `Install Packages...`
- Type in `AMR` and press Install
-
Install using [RStudio](http://www.rstudio.com) (recommended):
- Click on `Tools` and then `Install Packages...`
- Type in `AMR` and press Install
-  Install in R directly:
- `install.packages("AMR")`
### Install from GitHub (latest development version)
[](https://travis-ci.org/msberends/AMR)
[](https://github.com/msberends/AMR/commits/master)
[](https://github.com/msberends/AMR/commits/master)
[](https://codecov.io/gh/msberends/AMR)
```r
install.packages("devtools")
devtools::install_github("msberends/AMR")
```
## How to use it?
```r
# Call it with:
library(AMR)
# For a list of functions:
help(package = "AMR")
```
### Overwrite/force resistance based on EUCAST rules
This is also called *interpretive reading*.
```r
before <- data.frame(bactid = c("STAAUR", # Staphylococcus aureus
"ENCFAE", # Enterococcus faecalis
"ESCCOL", # Escherichia coli
"KLEPNE", # Klebsiella pneumoniae
"PSEAER"), # Pseudomonas aeruginosa
vanc = "-", # Vancomycin
amox = "-", # Amoxicillin
coli = "-", # Colistin
cfta = "-", # Ceftazidime
cfur = "-", # Cefuroxime
stringsAsFactors = FALSE)
before
# bactid vanc amox coli cfta cfur
# 1 STAAUR - - - - -
# 2 ENCFAE - - - - -
# 3 ESCCOL - - - - -
# 4 KLEPNE - - - - -
# 5 PSEAER - - - - -
# Now apply those rules; just need a column with bacteria ID's and antibiotic results:
after <- EUCAST_rules(before)
after
# bactid vanc amox coli cfta cfur
# 1 STAAUR - - R R -
# 2 ENCFAE - - R R R
# 3 ESCCOL R - - - -
# 4 KLEPNE R R - - -
# 5 PSEAER R R - - R
```
Bacteria ID's can be retrieved with the `guess_bactid` function. It uses any type of info about a microorganism as input. For example, all these will return value `STAAUR`, the ID of *S. aureus*:
```r
guess_bactid("stau")
guess_bactid("STAU")
guess_bactid("staaur")
guess_bactid("S. aureus")
guess_bactid("S aureus")
guess_bactid("Staphylococcus aureus")
guess_bactid("MRSA") # Methicillin Resistant S. aureus
guess_bactid("VISA") # Vancomycin Intermediate S. aureus
guess_bactid("VRSA") # Vancomycin Resistant S. aureus
```
### New classes
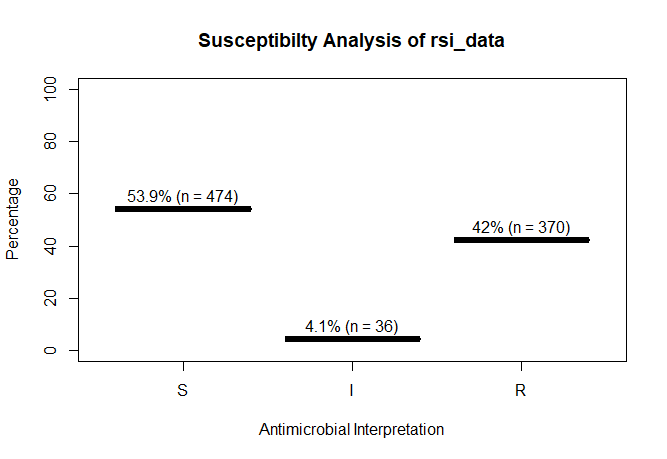
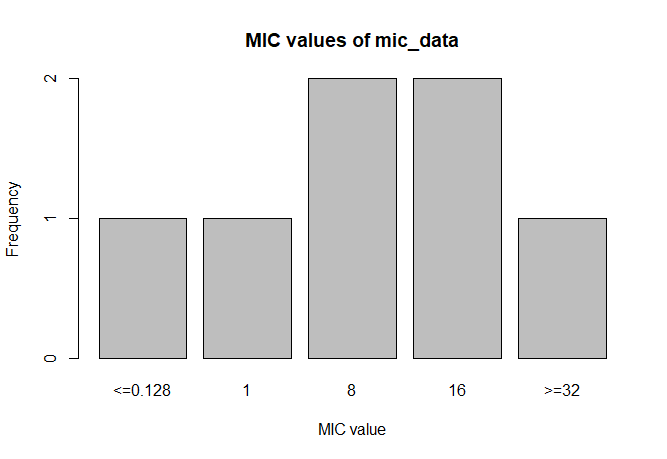
This package contains two new S3 classes: `mic` for MIC values (e.g. from Vitek or Phoenix) and `rsi` for antimicrobial drug interpretations (i.e. S, I and R). Both are actually ordered factors under the hood (an MIC of `2` being higher than `<=1` but lower than `>=32`, and for class `rsi` factors are ordered as `S < I < R`).
Both classes have extensions for existing generic functions like `print`, `summary` and `plot`.
```r
# Transform values to new classes
mic_data <- as.mic(c(">=32", "1.0", "8", "<=0.128", "8", "16", "16"))
rsi_data <- as.rsi(c(rep("S", 474), rep("I", 36), rep("R", 370)))
```
These functions also try to coerce valid values.
Quick overviews when just printing objects:
```r
mic_data
# Class 'mic': 7 isolates
#
# 0
#
# <=0.128 1 8 16 >=32
# 1 1 2 2 1
rsi_data
# Class 'rsi': 880 isolates
#
# : 0
# Sum of S: 474
# Sum of IR: 406
# - Sum of R: 370
# - Sum of I: 36
#
# %S %IR %I %R
# 53.9 46.1 4.1 42.0
```
A plot of `rsi_data`:
```r
plot(rsi_data)
```
A plot of `mic_data` (defaults to bar plot):
```r
plot(mic_data)
```
Other epidemiological functions:
```r
# Determine key antibiotic based on bacteria ID
key_antibiotics(...)
# Selection of first isolates of any patient
first_isolate(...)
# Calculate resistance levels of antibiotics, can be used with `summarise` (dplyr)
rsi(...)
# Predict resistance levels of antibiotics
rsi_predict(...)
# Get name of antibiotic by ATC code
abname(...)
abname("J01CR02", from = "atc", to = "umcg") # "AMCL"
```
### Frequency tables
Base R lacks a simple function to create frequency tables. We created such a function that works with almost all data types: `freq` (or `frequency_tbl`). It can be used in two ways:
```r
# Like base R:
freq(mydata$myvariable)
# And like tidyverse:
mydata %>% freq(myvariable)
```
Factors sort on item by default:
```r
septic_patients %>% freq(hospital_id)
# Frequency table of `hospital_id`
# Class: factor
# Length: 2000 (of which NA: 0 = 0.0%)
# Unique: 4
#
# Item Count Percent Cum. Count Cum. Percent (Factor Level)
# --- ----- ------ -------- ----------- ------------- ---------------
# 1 A 319 16.0% 319 16.0% 1
# 2 B 661 33.1% 980 49.0% 2
# 3 C 256 12.8% 1236 61.8% 3
# 4 D 764 38.2% 2000 100.0% 4
```
This can be changed with the `sort.count` parameter:
```r
septic_patients %>% freq(hospital_id, sort.count = TRUE)
# Frequency table of `hospital_id`
# Class: factor
# Length: 2000 (of which NA: 0 = 0.0%)
# Unique: 4
#
# Item Count Percent Cum. Count Cum. Percent (Factor Level)
# --- ----- ------ -------- ----------- ------------- ---------------
# 1 D 764 38.2% 764 38.2% 4
# 2 B 661 33.1% 1425 71.2% 2
# 3 A 319 16.0% 1744 87.2% 1
# 4 C 256 12.8% 2000 100.0% 3
```
All other types, like numbers, characters and dates, sort on count by default:
```r
septic_patients %>% freq(date)
# Frequency table of `date`
# Class: Date
# Length: 2000 (of which NA: 0 = 0.0%)
# Unique: 1151
#
# Oldest: 2 January 2002
# Newest: 28 December 2017 (+5839)
# Median: 7 Augustus 2009 (~48%)
#
# Item Count Percent Cum. Count Cum. Percent
# --- ----------- ------ -------- ----------- -------------
# 1 2016-05-21 10 0.5% 10 0.5%
# 2 2004-11-15 8 0.4% 18 0.9%
# 3 2013-07-29 8 0.4% 26 1.3%
# 4 2017-06-12 8 0.4% 34 1.7%
# 5 2015-11-19 7 0.4% 41 2.1%
# 6 2005-12-22 6 0.3% 47 2.4%
# 7 2015-10-12 6 0.3% 53 2.6%
# 8 2002-05-16 5 0.2% 58 2.9%
# 9 2004-02-02 5 0.2% 63 3.1%
# 10 2004-02-18 5 0.2% 68 3.4%
# 11 2005-08-16 5 0.2% 73 3.6%
# 12 2005-09-01 5 0.2% 78 3.9%
# 13 2006-06-29 5 0.2% 83 4.2%
# 14 2007-08-10 5 0.2% 88 4.4%
# 15 2008-08-29 5 0.2% 93 4.7%
# [ reached getOption("max.print.freq") -- omitted 1136 entries, n = 1907 (95.3%) ]
```
For numeric values, some extra descriptive statistics will be calculated:
```r
freq(runif(n = 10, min = 1, max = 5))
# Frequency table
# Class: numeric
# Length: 10 (of which NA: 0 = 0.0%)
# Unique: 10
#
# Mean: 3.4
# Std. dev.: 1.3 (CV: 0.38, MAD: 1.3)
# Five-Num: 1.6 | 2.0 | 3.9 | 4.7 | 4.8 (IQR: 2.7, CQV: 0.4)
# Outliers: 0
#
# Item Count Percent Cum. Count Cum. Percent
# --- --------- ------ -------- ----------- -------------
# 1 1.568997 1 10.0% 1 10.0%
# 2 1.993575 1 10.0% 2 20.0%
# 3 2.022348 1 10.0% 3 30.0%
# 4 2.236038 1 10.0% 4 40.0%
# 5 3.579828 1 10.0% 5 50.0%
# 6 4.178081 1 10.0% 6 60.0%
# 7 4.394818 1 10.0% 7 70.0%
# 8 4.689871 1 10.0% 8 80.0%
# 9 4.698626 1 10.0% 9 90.0%
# 10 4.751488 1 10.0% 10 100.0%
#
# Warning message:
# All observations are unique.
```
Learn more about this function with:
```r
?freq
```
### Databases included in package
Datasets to work with antibiotics and bacteria properties.
```r
# Dataset with 2000 random blood culture isolates from anonymised
# septic patients between 2001 and 2017 in 5 Dutch hospitals
septic_patients # A tibble: 2,000 x 49
# Dataset with ATC antibiotics codes, official names, trade names
# and DDD's (oral and parenteral)
antibiotics # A tibble: 420 x 18
# Dataset with bacteria codes and properties like gram stain and
# aerobic/anaerobic
microorganisms # A tibble: 2,453 x 12
```
## Copyright
[](https://github.com/msberends/AMR/blob/master/LICENSE)
This R package is licensed under the [GNU General Public License (GPL) v2.0](https://github.com/msberends/AMR/blob/master/LICENSE). In a nutshell, this means that this package:
- May be used for commercial purposes
- May be used for private purposes
- May **not** be used for patent purposes
- May be modified, although:
- Modifications **must** be released under the same license when distributing the package
- Changes made to the code **must** be documented
- May be distributed, although:
- Source code **must** be made available when the package is distributed
- A copy of the license and copyright notice **must** be included with the package.
- Comes with a LIMITATION of liability
- Comes with NO warranty
Install in R directly:
- `install.packages("AMR")`
### Install from GitHub (latest development version)
[](https://travis-ci.org/msberends/AMR)
[](https://github.com/msberends/AMR/commits/master)
[](https://github.com/msberends/AMR/commits/master)
[](https://codecov.io/gh/msberends/AMR)
```r
install.packages("devtools")
devtools::install_github("msberends/AMR")
```
## How to use it?
```r
# Call it with:
library(AMR)
# For a list of functions:
help(package = "AMR")
```
### Overwrite/force resistance based on EUCAST rules
This is also called *interpretive reading*.
```r
before <- data.frame(bactid = c("STAAUR", # Staphylococcus aureus
"ENCFAE", # Enterococcus faecalis
"ESCCOL", # Escherichia coli
"KLEPNE", # Klebsiella pneumoniae
"PSEAER"), # Pseudomonas aeruginosa
vanc = "-", # Vancomycin
amox = "-", # Amoxicillin
coli = "-", # Colistin
cfta = "-", # Ceftazidime
cfur = "-", # Cefuroxime
stringsAsFactors = FALSE)
before
# bactid vanc amox coli cfta cfur
# 1 STAAUR - - - - -
# 2 ENCFAE - - - - -
# 3 ESCCOL - - - - -
# 4 KLEPNE - - - - -
# 5 PSEAER - - - - -
# Now apply those rules; just need a column with bacteria ID's and antibiotic results:
after <- EUCAST_rules(before)
after
# bactid vanc amox coli cfta cfur
# 1 STAAUR - - R R -
# 2 ENCFAE - - R R R
# 3 ESCCOL R - - - -
# 4 KLEPNE R R - - -
# 5 PSEAER R R - - R
```
Bacteria ID's can be retrieved with the `guess_bactid` function. It uses any type of info about a microorganism as input. For example, all these will return value `STAAUR`, the ID of *S. aureus*:
```r
guess_bactid("stau")
guess_bactid("STAU")
guess_bactid("staaur")
guess_bactid("S. aureus")
guess_bactid("S aureus")
guess_bactid("Staphylococcus aureus")
guess_bactid("MRSA") # Methicillin Resistant S. aureus
guess_bactid("VISA") # Vancomycin Intermediate S. aureus
guess_bactid("VRSA") # Vancomycin Resistant S. aureus
```
### New classes
This package contains two new S3 classes: `mic` for MIC values (e.g. from Vitek or Phoenix) and `rsi` for antimicrobial drug interpretations (i.e. S, I and R). Both are actually ordered factors under the hood (an MIC of `2` being higher than `<=1` but lower than `>=32`, and for class `rsi` factors are ordered as `S < I < R`).
Both classes have extensions for existing generic functions like `print`, `summary` and `plot`.
```r
# Transform values to new classes
mic_data <- as.mic(c(">=32", "1.0", "8", "<=0.128", "8", "16", "16"))
rsi_data <- as.rsi(c(rep("S", 474), rep("I", 36), rep("R", 370)))
```
These functions also try to coerce valid values.
Quick overviews when just printing objects:
```r
mic_data
# Class 'mic': 7 isolates
#
# 0
#
# <=0.128 1 8 16 >=32
# 1 1 2 2 1
rsi_data
# Class 'rsi': 880 isolates
#
# : 0
# Sum of S: 474
# Sum of IR: 406
# - Sum of R: 370
# - Sum of I: 36
#
# %S %IR %I %R
# 53.9 46.1 4.1 42.0
```
A plot of `rsi_data`:
```r
plot(rsi_data)
```

A plot of `mic_data` (defaults to bar plot):
```r
plot(mic_data)
```

Other epidemiological functions:
```r
# Determine key antibiotic based on bacteria ID
key_antibiotics(...)
# Selection of first isolates of any patient
first_isolate(...)
# Calculate resistance levels of antibiotics, can be used with `summarise` (dplyr)
rsi(...)
# Predict resistance levels of antibiotics
rsi_predict(...)
# Get name of antibiotic by ATC code
abname(...)
abname("J01CR02", from = "atc", to = "umcg") # "AMCL"
```
### Frequency tables
Base R lacks a simple function to create frequency tables. We created such a function that works with almost all data types: `freq` (or `frequency_tbl`). It can be used in two ways:
```r
# Like base R:
freq(mydata$myvariable)
# And like tidyverse:
mydata %>% freq(myvariable)
```
Factors sort on item by default:
```r
septic_patients %>% freq(hospital_id)
# Frequency table of `hospital_id`
# Class: factor
# Length: 2000 (of which NA: 0 = 0.0%)
# Unique: 4
#
# Item Count Percent Cum. Count Cum. Percent (Factor Level)
# --- ----- ------ -------- ----------- ------------- ---------------
# 1 A 319 16.0% 319 16.0% 1
# 2 B 661 33.1% 980 49.0% 2
# 3 C 256 12.8% 1236 61.8% 3
# 4 D 764 38.2% 2000 100.0% 4
```
This can be changed with the `sort.count` parameter:
```r
septic_patients %>% freq(hospital_id, sort.count = TRUE)
# Frequency table of `hospital_id`
# Class: factor
# Length: 2000 (of which NA: 0 = 0.0%)
# Unique: 4
#
# Item Count Percent Cum. Count Cum. Percent (Factor Level)
# --- ----- ------ -------- ----------- ------------- ---------------
# 1 D 764 38.2% 764 38.2% 4
# 2 B 661 33.1% 1425 71.2% 2
# 3 A 319 16.0% 1744 87.2% 1
# 4 C 256 12.8% 2000 100.0% 3
```
All other types, like numbers, characters and dates, sort on count by default:
```r
septic_patients %>% freq(date)
# Frequency table of `date`
# Class: Date
# Length: 2000 (of which NA: 0 = 0.0%)
# Unique: 1151
#
# Oldest: 2 January 2002
# Newest: 28 December 2017 (+5839)
# Median: 7 Augustus 2009 (~48%)
#
# Item Count Percent Cum. Count Cum. Percent
# --- ----------- ------ -------- ----------- -------------
# 1 2016-05-21 10 0.5% 10 0.5%
# 2 2004-11-15 8 0.4% 18 0.9%
# 3 2013-07-29 8 0.4% 26 1.3%
# 4 2017-06-12 8 0.4% 34 1.7%
# 5 2015-11-19 7 0.4% 41 2.1%
# 6 2005-12-22 6 0.3% 47 2.4%
# 7 2015-10-12 6 0.3% 53 2.6%
# 8 2002-05-16 5 0.2% 58 2.9%
# 9 2004-02-02 5 0.2% 63 3.1%
# 10 2004-02-18 5 0.2% 68 3.4%
# 11 2005-08-16 5 0.2% 73 3.6%
# 12 2005-09-01 5 0.2% 78 3.9%
# 13 2006-06-29 5 0.2% 83 4.2%
# 14 2007-08-10 5 0.2% 88 4.4%
# 15 2008-08-29 5 0.2% 93 4.7%
# [ reached getOption("max.print.freq") -- omitted 1136 entries, n = 1907 (95.3%) ]
```
For numeric values, some extra descriptive statistics will be calculated:
```r
freq(runif(n = 10, min = 1, max = 5))
# Frequency table
# Class: numeric
# Length: 10 (of which NA: 0 = 0.0%)
# Unique: 10
#
# Mean: 3.4
# Std. dev.: 1.3 (CV: 0.38, MAD: 1.3)
# Five-Num: 1.6 | 2.0 | 3.9 | 4.7 | 4.8 (IQR: 2.7, CQV: 0.4)
# Outliers: 0
#
# Item Count Percent Cum. Count Cum. Percent
# --- --------- ------ -------- ----------- -------------
# 1 1.568997 1 10.0% 1 10.0%
# 2 1.993575 1 10.0% 2 20.0%
# 3 2.022348 1 10.0% 3 30.0%
# 4 2.236038 1 10.0% 4 40.0%
# 5 3.579828 1 10.0% 5 50.0%
# 6 4.178081 1 10.0% 6 60.0%
# 7 4.394818 1 10.0% 7 70.0%
# 8 4.689871 1 10.0% 8 80.0%
# 9 4.698626 1 10.0% 9 90.0%
# 10 4.751488 1 10.0% 10 100.0%
#
# Warning message:
# All observations are unique.
```
Learn more about this function with:
```r
?freq
```
### Databases included in package
Datasets to work with antibiotics and bacteria properties.
```r
# Dataset with 2000 random blood culture isolates from anonymised
# septic patients between 2001 and 2017 in 5 Dutch hospitals
septic_patients # A tibble: 2,000 x 49
# Dataset with ATC antibiotics codes, official names, trade names
# and DDD's (oral and parenteral)
antibiotics # A tibble: 420 x 18
# Dataset with bacteria codes and properties like gram stain and
# aerobic/anaerobic
microorganisms # A tibble: 2,453 x 12
```
## Copyright
[](https://github.com/msberends/AMR/blob/master/LICENSE)
This R package is licensed under the [GNU General Public License (GPL) v2.0](https://github.com/msberends/AMR/blob/master/LICENSE). In a nutshell, this means that this package:
- May be used for commercial purposes
- May be used for private purposes
- May **not** be used for patent purposes
- May be modified, although:
- Modifications **must** be released under the same license when distributing the package
- Changes made to the code **must** be documented
- May be distributed, although:
- Source code **must** be made available when the package is distributed
- A copy of the license and copyright notice **must** be included with the package.
- Comes with a LIMITATION of liability
- Comes with NO warranty