# `AMR`  ### An [R package](https://www.r-project.org) to simplify the analysis and prediction of Antimicrobial Resistance (AMR) and work with antibiotic properties by using evidence-based methods.
This R package was created for academic research by PhD students of the Faculty of Medical Sciences of the [University of Groningen](https://www.rug.nl) and the Medical Microbiology & Infection Prevention (MMBI) department of the [University Medical Center Groningen (UMCG)](https://www.umcg.nl).
:arrow_forward: Get it with `install.packages("AMR")` or see below for other possibilities. Read all changes and new functions in **[NEWS.md](https://github.com/msberends/AMR/blob/master/NEWS.md)**.
## Authors
### An [R package](https://www.r-project.org) to simplify the analysis and prediction of Antimicrobial Resistance (AMR) and work with antibiotic properties by using evidence-based methods.
This R package was created for academic research by PhD students of the Faculty of Medical Sciences of the [University of Groningen](https://www.rug.nl) and the Medical Microbiology & Infection Prevention (MMBI) department of the [University Medical Center Groningen (UMCG)](https://www.umcg.nl).
:arrow_forward: Get it with `install.packages("AMR")` or see below for other possibilities. Read all changes and new functions in **[NEWS.md](https://github.com/msberends/AMR/blob/master/NEWS.md)**.
## Authors
 Matthijs S. Berends1,2,a,
Christian F. Luz1,a,
Erwin E.A. Hassing2,
Corinna Glasner1,b,
Alex W. Friedrich1,b,
Bhanu Sinha1,b
1 Department of Medical Microbiology, University of Groningen, University Medical Center Groningen, Groningen, the Netherlands - [rug.nl](http://www.rug.nl) [umcg.nl](http://www.umcg.nl)
Matthijs S. Berends1,2,a,
Christian F. Luz1,a,
Erwin E.A. Hassing2,
Corinna Glasner1,b,
Alex W. Friedrich1,b,
Bhanu Sinha1,b
1 Department of Medical Microbiology, University of Groningen, University Medical Center Groningen, Groningen, the Netherlands - [rug.nl](http://www.rug.nl) [umcg.nl](http://www.umcg.nl)
2 Certe Medical Diagnostics & Advice, Groningen, the Netherlands - [certe.nl](http://www.certe.nl)
a R package author and thesis dissertant
b Thesis advisor




 ## Contents
* [Why this package?](#why-this-package)
* [ITIS](#itis)
* [How to get it?](#how-to-get-it)
* [Install from CRAN](#install-from-cran)
* [Install from Zenodo](#install-from-zenodo)
* [Install from GitHub](#install-from-github)
* [How to use it?](#how-to-use-it)
* [New classes](#new-classes)
* [Overwrite/force resistance based on EUCAST rules](#overwriteforce-resistance-based-on-eucast-rules)
* [Other (microbial) epidemiological functions](#other-microbial-epidemiological-functions)
* [Frequency tables](#frequency-tables)
* [Data sets included in package](#data-sets-included-in-package)
* [Benchmarks](#benchmarks)
* [Copyright](#copyright)
## Why this package?
This R package was intended **to make microbial epidemiology easier**. Most functions contain extensive help pages to get started.
The `AMR` package basically does four important things:
1. It **cleanses existing data**, by transforming it to reproducible and profound *classes*, making the most efficient use of R. These functions all use artificial intelligence to guess results that you would expect:
* Use `as.mo` to get an ID of a microorganism. The IDs are human readable for the trained eye - the ID of *Klebsiella pneumoniae* is "B_KLBSL_PNE" (B stands for Bacteria) and the ID of *S. aureus* is "B_STPHY_AUR". The function takes almost any text as input that looks like the name or code of a microorganism like "E. coli", "esco" and "esccol". Even `as.mo("MRSA")` will return the ID of *S. aureus*. Moreover, it can group all coagulase negative and positive *Staphylococci*, and can transform *Streptococci* into Lancefield groups. To find bacteria based on your input, it uses Artificial Intelligence to look up values in the included ITIS data, consisting of more than 18,000 microorganisms.
* Use `as.rsi` to transform values to valid antimicrobial results. It produces just S, I or R based on your input and warns about invalid values. Even values like "<=0.002; S" (combined MIC/RSI) will result in "S".
* Use `as.mic` to cleanse your MIC values. It produces a so-called factor (called *ordinal* in SPSS) with valid MIC values as levels. A value like "<=0.002; S" (combined MIC/RSI) will result in "<=0.002".
* Use `as.atc` to get the ATC code of an antibiotic as defined by the WHO. This package contains a database with most LIS codes, official names, DDDs and even trade names of antibiotics. For example, the values "Furabid", "Furadantin", "nitro" all return the ATC code of Nitrofurantoine.
2. It **enhances existing data** and **adds new data** from data sets included in this package.
* Use `EUCAST_rules` to apply [EUCAST expert rules to isolates](http://www.eucast.org/expert_rules_and_intrinsic_resistance/).
* Use `first_isolate` to identify the first isolates of every patient [using guidelines from the CLSI](https://clsi.org/standards/products/microbiology/documents/m39/) (Clinical and Laboratory Standards Institute).
* You can also identify first *weighted* isolates of every patient, an adjusted version of the CLSI guideline. This takes into account key antibiotics of every strain and compares them.
* Use `MDRO` (abbreviation of Multi Drug Resistant Organisms) to check your isolates for exceptional resistance with country-specific guidelines or EUCAST rules. Currently, national guidelines for Germany and the Netherlands are supported.
* The data set `microorganisms` contains the complete taxonomic tree of more than 18,000 microorganisms (bacteria, fungi/yeasts and protozoa). Furthermore, the colloquial name and Gram stain are available, which enables resistance analysis of e.g. different antibiotics per Gram stain. The package also contains functions to look up values in this data set like `mo_genus`, `mo_family`, `mo_gramstain` or even `mo_phylum`. As they use `as.mo` internally, they also use artificial intelligence. For example, `mo_genus("MRSA")` and `mo_genus("S. aureus")` will both return `"Staphylococcus"`. They also come with support for German, Dutch, French, Italian, Spanish and Portuguese. These functions can be used to add new variables to your data.
* The data set `antibiotics` contains the ATC code, LIS codes, official name, trivial name and DDD of both oral and parenteral administration. It also contains a total of 298 trade names. Use functions like `ab_name` and `ab_tradenames` to look up values. The `ab_*` functions use `as.atc` internally so they support AI to guess your expected result. For example, `ab_name("Fluclox")`, `ab_name("Floxapen")` and `ab_name("J01CF05")` will all return `"Flucloxacillin"`. These functions can again be used to add new variables to your data.
3. It **analyses the data** with convenient functions that use well-known methods.
* Calculate the resistance (and even co-resistance) of microbial isolates with the `portion_R`, `portion_IR`, `portion_I`, `portion_SI` and `portion_S` functions. Similarly, the *amount* of isolates can be determined with the `count_R`, `count_IR`, `count_I`, `count_SI` and `count_S` functions. All these functions can be used [with the `dplyr` package](https://dplyr.tidyverse.org/#usage) (e.g. in conjunction with [`summarise`](https://dplyr.tidyverse.org/reference/summarise.html))
* Plot AMR results with `geom_rsi`, a function made for the `ggplot2` package
* Predict antimicrobial resistance for the nextcoming years using logistic regression models with the `resistance_predict` function
* Conduct descriptive statistics to enhance base R: calculate kurtosis, skewness and create frequency tables
4. It **teaches the user** how to use all the above actions.
* The package contains extensive help pages with many examples.
* It also contains an example data set called `septic_patients`. This data set contains:
* 2,000 blood culture isolates from anonymised septic patients between 2001 and 2017 in the Northern Netherlands
* Results of 40 antibiotics (each antibiotic in its own column) with a total of 38,414 antimicrobial results
* Real and genuine data
### ITIS
## Contents
* [Why this package?](#why-this-package)
* [ITIS](#itis)
* [How to get it?](#how-to-get-it)
* [Install from CRAN](#install-from-cran)
* [Install from Zenodo](#install-from-zenodo)
* [Install from GitHub](#install-from-github)
* [How to use it?](#how-to-use-it)
* [New classes](#new-classes)
* [Overwrite/force resistance based on EUCAST rules](#overwriteforce-resistance-based-on-eucast-rules)
* [Other (microbial) epidemiological functions](#other-microbial-epidemiological-functions)
* [Frequency tables](#frequency-tables)
* [Data sets included in package](#data-sets-included-in-package)
* [Benchmarks](#benchmarks)
* [Copyright](#copyright)
## Why this package?
This R package was intended **to make microbial epidemiology easier**. Most functions contain extensive help pages to get started.
The `AMR` package basically does four important things:
1. It **cleanses existing data**, by transforming it to reproducible and profound *classes*, making the most efficient use of R. These functions all use artificial intelligence to guess results that you would expect:
* Use `as.mo` to get an ID of a microorganism. The IDs are human readable for the trained eye - the ID of *Klebsiella pneumoniae* is "B_KLBSL_PNE" (B stands for Bacteria) and the ID of *S. aureus* is "B_STPHY_AUR". The function takes almost any text as input that looks like the name or code of a microorganism like "E. coli", "esco" and "esccol". Even `as.mo("MRSA")` will return the ID of *S. aureus*. Moreover, it can group all coagulase negative and positive *Staphylococci*, and can transform *Streptococci* into Lancefield groups. To find bacteria based on your input, it uses Artificial Intelligence to look up values in the included ITIS data, consisting of more than 18,000 microorganisms.
* Use `as.rsi` to transform values to valid antimicrobial results. It produces just S, I or R based on your input and warns about invalid values. Even values like "<=0.002; S" (combined MIC/RSI) will result in "S".
* Use `as.mic` to cleanse your MIC values. It produces a so-called factor (called *ordinal* in SPSS) with valid MIC values as levels. A value like "<=0.002; S" (combined MIC/RSI) will result in "<=0.002".
* Use `as.atc` to get the ATC code of an antibiotic as defined by the WHO. This package contains a database with most LIS codes, official names, DDDs and even trade names of antibiotics. For example, the values "Furabid", "Furadantin", "nitro" all return the ATC code of Nitrofurantoine.
2. It **enhances existing data** and **adds new data** from data sets included in this package.
* Use `EUCAST_rules` to apply [EUCAST expert rules to isolates](http://www.eucast.org/expert_rules_and_intrinsic_resistance/).
* Use `first_isolate` to identify the first isolates of every patient [using guidelines from the CLSI](https://clsi.org/standards/products/microbiology/documents/m39/) (Clinical and Laboratory Standards Institute).
* You can also identify first *weighted* isolates of every patient, an adjusted version of the CLSI guideline. This takes into account key antibiotics of every strain and compares them.
* Use `MDRO` (abbreviation of Multi Drug Resistant Organisms) to check your isolates for exceptional resistance with country-specific guidelines or EUCAST rules. Currently, national guidelines for Germany and the Netherlands are supported.
* The data set `microorganisms` contains the complete taxonomic tree of more than 18,000 microorganisms (bacteria, fungi/yeasts and protozoa). Furthermore, the colloquial name and Gram stain are available, which enables resistance analysis of e.g. different antibiotics per Gram stain. The package also contains functions to look up values in this data set like `mo_genus`, `mo_family`, `mo_gramstain` or even `mo_phylum`. As they use `as.mo` internally, they also use artificial intelligence. For example, `mo_genus("MRSA")` and `mo_genus("S. aureus")` will both return `"Staphylococcus"`. They also come with support for German, Dutch, French, Italian, Spanish and Portuguese. These functions can be used to add new variables to your data.
* The data set `antibiotics` contains the ATC code, LIS codes, official name, trivial name and DDD of both oral and parenteral administration. It also contains a total of 298 trade names. Use functions like `ab_name` and `ab_tradenames` to look up values. The `ab_*` functions use `as.atc` internally so they support AI to guess your expected result. For example, `ab_name("Fluclox")`, `ab_name("Floxapen")` and `ab_name("J01CF05")` will all return `"Flucloxacillin"`. These functions can again be used to add new variables to your data.
3. It **analyses the data** with convenient functions that use well-known methods.
* Calculate the resistance (and even co-resistance) of microbial isolates with the `portion_R`, `portion_IR`, `portion_I`, `portion_SI` and `portion_S` functions. Similarly, the *amount* of isolates can be determined with the `count_R`, `count_IR`, `count_I`, `count_SI` and `count_S` functions. All these functions can be used [with the `dplyr` package](https://dplyr.tidyverse.org/#usage) (e.g. in conjunction with [`summarise`](https://dplyr.tidyverse.org/reference/summarise.html))
* Plot AMR results with `geom_rsi`, a function made for the `ggplot2` package
* Predict antimicrobial resistance for the nextcoming years using logistic regression models with the `resistance_predict` function
* Conduct descriptive statistics to enhance base R: calculate kurtosis, skewness and create frequency tables
4. It **teaches the user** how to use all the above actions.
* The package contains extensive help pages with many examples.
* It also contains an example data set called `septic_patients`. This data set contains:
* 2,000 blood culture isolates from anonymised septic patients between 2001 and 2017 in the Northern Netherlands
* Results of 40 antibiotics (each antibiotic in its own column) with a total of 38,414 antimicrobial results
* Real and genuine data
### ITIS
 This package contains the **complete microbial taxonomic data** (with all seven taxonomic ranks - from subkingdom to subspecies) from the publicly available Integrated Taxonomic Information System (ITIS, https://www.itis.gov).
All (sub)species from the taxonomic kingdoms Bacteria, Fungi and Protozoa are included in this package, as well as all previously accepted names known to ITIS. Furthermore, the responsible authors and year of publication are available. This allows users to use authoritative taxonomic information for their data analysis on any microorganism, not only human pathogens.
ITIS is a partnership of U.S., Canadian, and Mexican agencies and taxonomic specialists.
**Get a note when a species was renamed**
```r
mo_shortname("Chlamydia psittaci")
# Note: 'Chlamydia psittaci' (Page, 1968) was renamed 'Chlamydophila psittaci' (Everett et al., 1999)
# [1] "C. psittaci"
```
**Get any property from the entire taxonomic tree for all included species**
```r
mo_class("E. coli")
# [1] "Gammaproteobacteria"
mo_family("E. coli")
# [1] "Enterobacteriaceae"
mo_ref("E. coli")
# [1] "Castellani and Chalmers, 1919"
```
**Do not get mistaken - the package only includes microorganisms**
```r
mo_phylum("C. elegans")
# [1] "Cyanobacteria" # Bacteria?!
mo_fullname("C. elegans")
# [1] "Chroococcus limneticus elegans" # Because a microorganism was found
```
## How to get it?
All stable versions of this package [are published on CRAN](http://cran.r-project.org/package=AMR), the official R network with a peer-reviewed submission process.
### Install from CRAN
[](http://cran.r-project.org/package=AMR) [](http://cran.r-project.org/package=AMR)
(Note: Downloads measured only by [cran.rstudio.com](https://cran.rstudio.com/package=AMR), this excludes e.g. the official [cran.r-project.org](https://cran.r-project.org/package=AMR))
-
This package contains the **complete microbial taxonomic data** (with all seven taxonomic ranks - from subkingdom to subspecies) from the publicly available Integrated Taxonomic Information System (ITIS, https://www.itis.gov).
All (sub)species from the taxonomic kingdoms Bacteria, Fungi and Protozoa are included in this package, as well as all previously accepted names known to ITIS. Furthermore, the responsible authors and year of publication are available. This allows users to use authoritative taxonomic information for their data analysis on any microorganism, not only human pathogens.
ITIS is a partnership of U.S., Canadian, and Mexican agencies and taxonomic specialists.
**Get a note when a species was renamed**
```r
mo_shortname("Chlamydia psittaci")
# Note: 'Chlamydia psittaci' (Page, 1968) was renamed 'Chlamydophila psittaci' (Everett et al., 1999)
# [1] "C. psittaci"
```
**Get any property from the entire taxonomic tree for all included species**
```r
mo_class("E. coli")
# [1] "Gammaproteobacteria"
mo_family("E. coli")
# [1] "Enterobacteriaceae"
mo_ref("E. coli")
# [1] "Castellani and Chalmers, 1919"
```
**Do not get mistaken - the package only includes microorganisms**
```r
mo_phylum("C. elegans")
# [1] "Cyanobacteria" # Bacteria?!
mo_fullname("C. elegans")
# [1] "Chroococcus limneticus elegans" # Because a microorganism was found
```
## How to get it?
All stable versions of this package [are published on CRAN](http://cran.r-project.org/package=AMR), the official R network with a peer-reviewed submission process.
### Install from CRAN
[](http://cran.r-project.org/package=AMR) [](http://cran.r-project.org/package=AMR)
(Note: Downloads measured only by [cran.rstudio.com](https://cran.rstudio.com/package=AMR), this excludes e.g. the official [cran.r-project.org](https://cran.r-project.org/package=AMR))
-  Install using [RStudio](http://www.rstudio.com) (recommended):
- Click on `Tools` and then `Install Packages...`
- Type in `AMR` and press Install
-
Install using [RStudio](http://www.rstudio.com) (recommended):
- Click on `Tools` and then `Install Packages...`
- Type in `AMR` and press Install
-  Install in R directly:
- `install.packages("AMR")`
### Install from Zenodo
[](https://doi.org/10.5281/zenodo.1305355)
This package was also published on Zenodo (stable releases only): https://doi.org/10.5281/zenodo.1305355
### Install from GitHub
This is the latest **development version**. Although it may contain bugfixes and even new functions compared to the latest released version on CRAN, it is also subject to change and may be unstable or behave unexpectedly. Always consider this a beta version. All below 'badges' should be green:
Development Test | Result | Reference
--- | :---: | ---
All functions checked on Linux and macOS | [](https://travis-ci.org/msberends/AMR) | Travis CI, GmbH [[ref 1]](https://travis-ci.org/msberends/AMR)
All functions checked on Windows | [](https://ci.appveyor.com/project/msberends/AMR) | Appveyor Systems Inc. [[ref 2]](https://ci.appveyor.com/project/msberends/AMR)
Percentage of syntax lines checked | [](https://codecov.io/gh/msberends/AMR) | Codecov LLC [[ref 3]](https://codecov.io/gh/msberends/AMR)
If so, try it with:
```r
install.packages("devtools")
devtools::install_github("msberends/AMR")
```
## How to use it?
```r
# Call it with:
library(AMR)
# For a list of functions:
help(package = "AMR")
```
### New classes
This package contains two new S3 classes: `mic` for MIC values (e.g. from Vitek or Phoenix) and `rsi` for antimicrobial drug interpretations (i.e. S, I and R). Both are actually ordered factors under the hood (an MIC of `2` being higher than `<=1` but lower than `>=32`, and for class `rsi` factors are ordered as `S < I < R`).
Both classes have extensions for existing generic functions like `print`, `summary` and `plot`.
These functions also try to coerce valid values.
#### RSI
The `septic_patients` data set comes with antimicrobial results of more than 40 different drugs. For example, columns `amox` and `cipr` contain results of amoxicillin and ciprofloxacin, respectively.
```r
summary(septic_patients[, c("amox", "cipr")])
# amox cipr
# Mode :rsi Mode :rsi
# :1002 :596
# Sum S :336 Sum S :1108
# Sum IR:662 Sum IR:296
# -Sum R:659 -Sum R:227
# -Sum I:3 -Sum I:69
```
You can use the `plot` function from base R:
```r
plot(septic_patients$cipr)
```
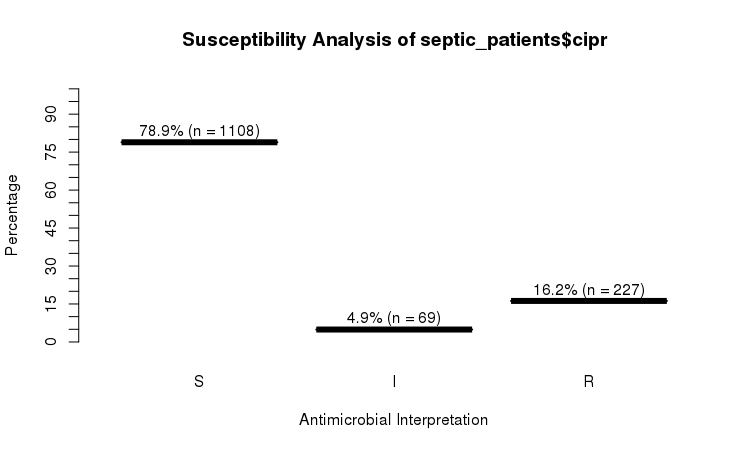
Or use the `ggplot2` and `dplyr` packages to create more appealing plots:
```r
library(dplyr)
library(ggplot2)
septic_patients %>%
select(amox, nitr, fosf, trim, cipr) %>%
ggplot_rsi()
```
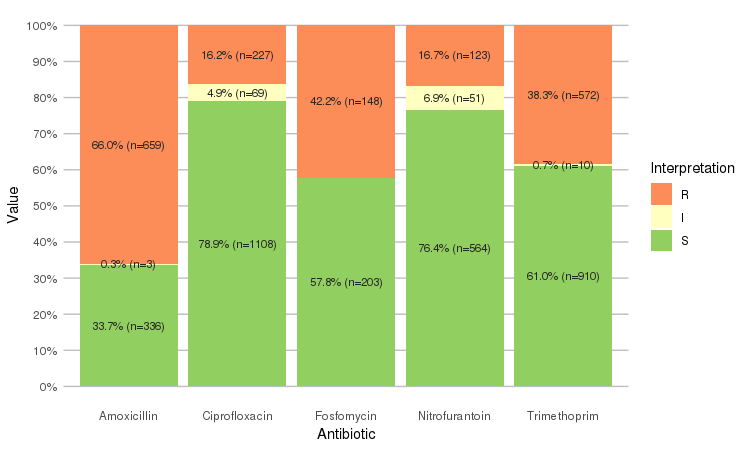
Adjust it with any parameter you know from the `ggplot2` package:
```r
septic_patients %>%
select(amox, nitr, fosf, trim, cipr) %>%
ggplot_rsi(datalabels = FALSE,
width = 0.5, colour = "black", size = 1, linetype = 2, alpha = 0.25)
```
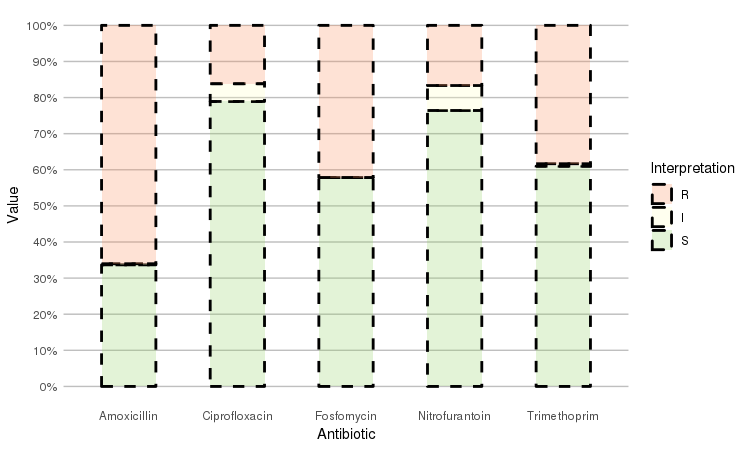
It also supports grouping variables. Let's say we want to compare resistance of drugs against Urine Tract Infections (UTI) between hospitals A to D (variable `hospital_id`):
```r
septic_patients %>%
select(hospital_id, amox, nitr, fosf, trim, cipr) %>%
group_by(hospital_id) %>%
ggplot_rsi(x = "hospital_id",
facet = "Antibiotic",
nrow = 1,
datalabels = FALSE) +
labs(title = "AMR of Anti-UTI Drugs Per Hospital",
x = "Hospital")
```
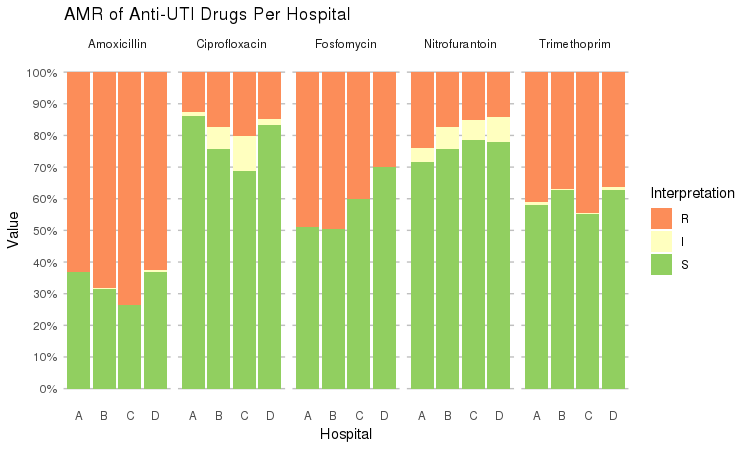
You could use this to group on anything in your plots: Gram stain, age (group), genus, geographic location, et cetera.
#### MIC
```r
# Transform values to new class
mic_data <- as.mic(c(">=32", "1.0", "8", "<=0.128", "8", "16", "16"))
summary(mic_data)
# Mode:mic
# :0
# Min.:<=0.128
# Max.:>=32
plot(mic_data)
```

### Overwrite/force resistance based on EUCAST rules
This is also called *interpretive reading*.
```r
before <- data.frame(bact = c("STAAUR", # Staphylococcus aureus
"ENCFAE", # Enterococcus faecalis
"ESCCOL", # Escherichia coli
"KLEPNE", # Klebsiella pneumoniae
"PSEAER"), # Pseudomonas aeruginosa
vanc = "-", # Vancomycin
amox = "-", # Amoxicillin
coli = "-", # Colistin
cfta = "-", # Ceftazidime
cfur = "-", # Cefuroxime
stringsAsFactors = FALSE)
before
# bact vanc amox coli cfta cfur
# 1 STAAUR - - - - -
# 2 ENCFAE - - - - -
# 3 ESCCOL - - - - -
# 4 KLEPNE - - - - -
# 5 PSEAER - - - - -
# Now apply those rules; just need a column with bacteria IDs and antibiotic results:
after <- EUCAST_rules(before, col_mo = "bact")
after
# bact vanc amox coli cfta cfur
# 1 STAAUR - - R R -
# 2 ENCFAE - - R R R
# 3 ESCCOL R - - - -
# 4 KLEPNE R R - - -
# 5 PSEAER R R - - R
```
Bacteria IDs can be retrieved with the `guess_mo` function. It uses any type of info about a microorganism as input. For example, all these will return value `STAAUR`, the ID of *S. aureus*:
```r
guess_mo("stau")
guess_mo("STAU")
guess_mo("staaur")
guess_mo("S. aureus")
guess_mo("S aureus")
guess_mo("Staphylococcus aureus")
guess_mo("MRSA") # Methicillin Resistant S. aureus
guess_mo("VISA") # Vancomycin Intermediate S. aureus
guess_mo("VRSA") # Vancomycin Resistant S. aureus
```
### Other (microbial) epidemiological functions
```r
# G-test to replace Chi squared test
g.test(...)
# Determine key antibiotic based on bacteria ID
key_antibiotics(...)
# Selection of first isolates of any patient
first_isolate(...)
# Calculate resistance levels of antibiotics, can be used with `summarise` (dplyr)
rsi(...)
# Predict resistance levels of antibiotics
rsi_predict(...)
# Get name of antibiotic by ATC code
abname(...)
abname("J01CR02", from = "atc", to = "umcg") # "AMCL"
```
### Frequency tables
Base R lacks a simple function to create frequency tables. We created such a function that works with almost all data types: `freq` (or `frequency_tbl`). It can be used in two ways:
```r
# Like base R:
freq(mydata$myvariable)
# And like tidyverse:
mydata %>% freq(myvariable)
```
Factors sort on item by default:
```r
septic_patients %>% freq(hospital_id)
# Frequency table of `hospital_id`
# Class: factor
# Length: 2000 (of which NA: 0 = 0.0%)
# Unique: 4
#
# Item Count Percent Cum. Count Cum. Percent (Factor Level)
# --- ----- ------ -------- ----------- ------------- ---------------
# 1 A 319 16.0% 319 16.0% 1
# 2 B 661 33.1% 980 49.0% 2
# 3 C 256 12.8% 1236 61.8% 3
# 4 D 764 38.2% 2000 100.0% 4
```
This can be changed with the `sort.count` parameter:
```r
septic_patients %>% freq(hospital_id, sort.count = TRUE)
# Frequency table of `hospital_id`
# Class: factor
# Length: 2000 (of which NA: 0 = 0.0%)
# Unique: 4
#
# Item Count Percent Cum. Count Cum. Percent (Factor Level)
# --- ----- ------ -------- ----------- ------------- ---------------
# 1 D 764 38.2% 764 38.2% 4
# 2 B 661 33.1% 1425 71.2% 2
# 3 A 319 16.0% 1744 87.2% 1
# 4 C 256 12.8% 2000 100.0% 3
```
All other types, like numbers, characters and dates, sort on count by default:
```r
septic_patients %>% freq(date)
# Frequency table of `date`
# Class: Date
# Length: 2000 (of which NA: 0 = 0.0%)
# Unique: 1151
#
# Oldest: 2 January 2002
# Newest: 28 December 2017 (+5839)
# Median: 7 Augustus 2009 (~48%)
#
# Item Count Percent Cum. Count Cum. Percent
# --- ----------- ------ -------- ----------- -------------
# 1 2016-05-21 10 0.5% 10 0.5%
# 2 2004-11-15 8 0.4% 18 0.9%
# 3 2013-07-29 8 0.4% 26 1.3%
# 4 2017-06-12 8 0.4% 34 1.7%
# 5 2015-11-19 7 0.4% 41 2.1%
# 6 2005-12-22 6 0.3% 47 2.4%
# 7 2015-10-12 6 0.3% 53 2.6%
# 8 2002-05-16 5 0.2% 58 2.9%
# 9 2004-02-02 5 0.2% 63 3.1%
# 10 2004-02-18 5 0.2% 68 3.4%
# 11 2005-08-16 5 0.2% 73 3.6%
# 12 2005-09-01 5 0.2% 78 3.9%
# 13 2006-06-29 5 0.2% 83 4.2%
# 14 2007-08-10 5 0.2% 88 4.4%
# 15 2008-08-29 5 0.2% 93 4.7%
# [ reached getOption("max.print.freq") -- omitted 1136 entries, n = 1907 (95.3%) ]
```
For numeric values, some extra descriptive statistics will be calculated:
```r
freq(runif(n = 10, min = 1, max = 5))
# Frequency table
# Class: numeric
# Length: 10 (of which NA: 0 = 0.0%)
# Unique: 10
#
# Mean: 3.4
# Std. dev.: 1.3 (CV: 0.38, MAD: 1.3)
# Five-Num: 1.6 | 2.0 | 3.9 | 4.7 | 4.8 (IQR: 2.7, CQV: 0.4)
# Outliers: 0
#
# Item Count Percent Cum. Count Cum. Percent
# --- --------- ------ -------- ----------- -------------
# 1 1.568997 1 10.0% 1 10.0%
# 2 1.993575 1 10.0% 2 20.0%
# 3 2.022348 1 10.0% 3 30.0%
# 4 2.236038 1 10.0% 4 40.0%
# 5 3.579828 1 10.0% 5 50.0%
# 6 4.178081 1 10.0% 6 60.0%
# 7 4.394818 1 10.0% 7 70.0%
# 8 4.689871 1 10.0% 8 80.0%
# 9 4.698626 1 10.0% 9 90.0%
# 10 4.751488 1 10.0% 10 100.0%
#
# Warning message:
# All observations are unique.
```
Learn more about this function with:
```r
?freq
```
### Data sets included in package
Data sets to work with antibiotics and bacteria properties.
```r
# Data set with complete taxonomic trees from ITIS, containing of
# the three kingdoms Bacteria, Fungi and Protozoa
microorganisms # A tibble: 18,831 x 15
# Data set with 2000 random blood culture isolates from anonymised
# septic patients between 2001 and 2017 in 5 Dutch hospitals
septic_patients # A tibble: 2,000 x 49
# Data set with ATC antibiotics codes, official names, trade names
# and DDDs (oral and parenteral)
antibiotics # A tibble: 423 x 18
```
## Benchmarks
One of the most important features of this package is the complete microbial taxonomic database, supplied by ITIS (https://www.itis.gov). We created a function `as.mo` that transforms any user input value to a valid microbial ID by using AI (Artificial Intelligence) and based on the taxonomic tree of ITIS.
Using the `microbenchmark` package, we can review the calculation performance of this function.
```r
library(microbenchmark)
```
In the next test, we try to 'coerce' different input values for *Staphylococcus aureus*. The actual result is the same every time: it returns its MO code `B_STAPHY_AUR` (*B* stands for *Bacteria*, the taxonomic kingdom).
But the calculation time differs a lot. Here, the AI effect can be reviewed best:
```r
microbenchmark(A = as.mo("stau"),
B = as.mo("staaur"),
C = as.mo("S. aureus"),
D = as.mo("S. aureus"),
E = as.mo("STAAUR"),
F = as.mo("Staphylococcus aureus"),
times = 10,
unit = "ms")
# Unit: milliseconds
# expr min lq mean median uq max neval
# A 36.05088 36.14782 36.65635 36.24466 36.43075 39.78544 10
# B 16.43575 16.46885 16.67816 16.66053 16.84858 16.95299 10
# C 14.44150 14.52182 16.81197 14.59173 14.67854 36.75244 10
# D 14.49765 14.58153 16.71666 14.59414 14.61094 35.50731 10
# E 14.45212 14.75146 14.82033 14.85559 14.96433 15.03438 10
# F 10.69445 10.73852 10.80334 10.79596 10.86856 10.97465 10
```
The more an input value resembles a full name, the faster the result will be found. In the table above, all measurements are in milliseconds, tested on a quite regular Linux server from 2007 with 2 GB RAM. A value of 10.8 milliseconds means it can roughly determine 93 different input values per second. It case of 36.2 milliseconds, this is only 28 input values per second.
To improve speed, the `as.mo` function also takes into account the prevalence of human pathogenic microorganisms. The downside is of course that less prevalent microorganisms will be determined far less faster. See this example for the ID of *Burkholderia nodosa* (`B_BRKHL_NOD`):
```r
microbenchmark(B = as.mo("burnod"),
C = as.mo("B. nodosa"),
D = as.mo("B. nodosa"),
E = as.mo("BURNOD"),
F = as.mo("Burkholderia nodosa"),
times = 10,
unit = "ms")
# Unit: milliseconds
# expr min lq mean median uq max neval
# B 175.9446 176.80440 179.18240 177.00131 177.62021 198.86286 10
# C 88.1902 88.57705 89.08851 88.84293 89.15498 91.76621 10
# D 110.2641 110.67497 113.66290 111.20534 111.80744 134.44699 10
# E 175.0728 177.04235 207.83542 190.38109 200.33448 388.12177 10
# F 45.0778 45.31617 52.72430 45.62962 67.85262 70.42250 10
```
(Note: `A` is missing here, because `as.mo("buno")` returns `F_BUELL_NOT`: the ID of the fungus *Buellia notabilis*)
That takes up to 12 times as much time! A value of 190.4 milliseconds means it can only determine 5 different input values per second. We can conclude that looking up arbitrary codes of less prevalent microorganisms is the worst way to go, in terms of calculation performance.
To relieve this pitfall and further improve performance, two important calculations take almost no time at all: **repetive results** and **already precalculated results**.
Let's set up 25,000 entries of `"Staphylococcus aureus"` and check its speed:
```r
repetive_results <- rep("Staphylococcus aureus", 25000)
microbenchmark(A = as.mo(repetive_results),
times = 10,
unit = "ms")
# Unit: milliseconds
# expr min lq mean median uq max neval
# A 14.61282 14.6372 14.70817 14.72597 14.76124 14.78498 10
```
So transforming 25,000 times (!) `"Staphylococcus aureus"` only takes 4 ms (0.004 seconds) more than transforming it once. You only lose time on your unique input values.
What about precalculated results? This package also contains helper functions for specific microbial properties, for example `mo_fullname`. It returns the full microbial name (genus, species and possibly subspecies) and uses `as.mo` internally. If the input is however an already precalculated result, it almost doesn't take any time at all (see 'C' below):
```r
microbenchmark(A = mo_fullname("B_STPHY_AUR"),
B = mo_fullname("S. aureus"),
C = mo_fullname("Staphylococcus aureus"),
times = 10,
unit = "ms")
# Unit: milliseconds
# expr min lq mean median uq max neval
# A 13.548652 13.74588 13.8052969 13.813594 13.881165 14.090969 10
# B 15.079781 15.16785 15.3835842 15.374477 15.395115 16.072995 10
# C 0.171182 0.18563 0.2306307 0.203413 0.224610 0.492312 10
```
So going from `mo_fullname("Staphylococcus aureus")` to `"Staphylococcus aureus"` takes 0.0002 seconds - it doesn't even start calculating *if the result would be the same as the expected resulting value*. That goes for all helper functions:
```r
microbenchmark(A = mo_species("aureus"),
B = mo_genus("Staphylococcus"),
C = mo_fullname("Staphylococcus aureus"),
D = mo_family("Staphylococcaceae"),
E = mo_order("Bacillales"),
F = mo_class("Bacilli"),
G = mo_phylum("Firmicutes"),
H = mo_subkingdom("Posibacteria"),
times = 10,
unit = "ms")
# Unit: milliseconds
# expr min lq mean median uq max neval
# A 0.145270 0.158750 0.1908419 0.1693655 0.218255 0.300528 10
# B 0.182985 0.184522 0.2025408 0.1970235 0.209944 0.243328 10
# C 0.176280 0.201632 0.2618147 0.2303025 0.339499 0.388249 10
# D 0.136890 0.139054 0.1552231 0.1518010 0.168738 0.193042 10
# E 0.100921 0.116496 0.1321823 0.1222930 0.129976 0.230477 10
# F 0.103017 0.110281 0.1214480 0.1199880 0.124319 0.147506 10
# G 0.099246 0.110280 0.1195553 0.1188705 0.125436 0.149741 10
# H 0.114331 0.117264 0.1249819 0.1220830 0.129557 0.143385 10
```
Of course, when running `mo_phylum("Firmicutes")` the function has zero knowledge about the actual microorganism, namely *S. aureus*. But since the result would be `"Firmicutes"` too, there is no point in calculating the result. And since this package 'knows' all phyla of all known microorganisms (according to ITIS), it can just return the initial value immediately.
## Copyright
[](https://github.com/msberends/AMR/blob/master/LICENSE)
This R package is licensed under the [GNU General Public License (GPL) v2.0](https://github.com/msberends/AMR/blob/master/LICENSE). In a nutshell, this means that this package:
- May be used for commercial purposes
- May be used for private purposes
- May **not** be used for patent purposes
- May be modified, although:
- Modifications **must** be released under the same license when distributing the package
- Changes made to the code **must** be documented
- May be distributed, although:
- Source code **must** be made available when the package is distributed
- A copy of the license and copyright notice **must** be included with the package.
- Comes with a LIMITATION of liability
- Comes with NO warranty
Install in R directly:
- `install.packages("AMR")`
### Install from Zenodo
[](https://doi.org/10.5281/zenodo.1305355)
This package was also published on Zenodo (stable releases only): https://doi.org/10.5281/zenodo.1305355
### Install from GitHub
This is the latest **development version**. Although it may contain bugfixes and even new functions compared to the latest released version on CRAN, it is also subject to change and may be unstable or behave unexpectedly. Always consider this a beta version. All below 'badges' should be green:
Development Test | Result | Reference
--- | :---: | ---
All functions checked on Linux and macOS | [](https://travis-ci.org/msberends/AMR) | Travis CI, GmbH [[ref 1]](https://travis-ci.org/msberends/AMR)
All functions checked on Windows | [](https://ci.appveyor.com/project/msberends/AMR) | Appveyor Systems Inc. [[ref 2]](https://ci.appveyor.com/project/msberends/AMR)
Percentage of syntax lines checked | [](https://codecov.io/gh/msberends/AMR) | Codecov LLC [[ref 3]](https://codecov.io/gh/msberends/AMR)
If so, try it with:
```r
install.packages("devtools")
devtools::install_github("msberends/AMR")
```
## How to use it?
```r
# Call it with:
library(AMR)
# For a list of functions:
help(package = "AMR")
```
### New classes
This package contains two new S3 classes: `mic` for MIC values (e.g. from Vitek or Phoenix) and `rsi` for antimicrobial drug interpretations (i.e. S, I and R). Both are actually ordered factors under the hood (an MIC of `2` being higher than `<=1` but lower than `>=32`, and for class `rsi` factors are ordered as `S < I < R`).
Both classes have extensions for existing generic functions like `print`, `summary` and `plot`.
These functions also try to coerce valid values.
#### RSI
The `septic_patients` data set comes with antimicrobial results of more than 40 different drugs. For example, columns `amox` and `cipr` contain results of amoxicillin and ciprofloxacin, respectively.
```r
summary(septic_patients[, c("amox", "cipr")])
# amox cipr
# Mode :rsi Mode :rsi
# :1002 :596
# Sum S :336 Sum S :1108
# Sum IR:662 Sum IR:296
# -Sum R:659 -Sum R:227
# -Sum I:3 -Sum I:69
```
You can use the `plot` function from base R:
```r
plot(septic_patients$cipr)
```

Or use the `ggplot2` and `dplyr` packages to create more appealing plots:
```r
library(dplyr)
library(ggplot2)
septic_patients %>%
select(amox, nitr, fosf, trim, cipr) %>%
ggplot_rsi()
```

Adjust it with any parameter you know from the `ggplot2` package:
```r
septic_patients %>%
select(amox, nitr, fosf, trim, cipr) %>%
ggplot_rsi(datalabels = FALSE,
width = 0.5, colour = "black", size = 1, linetype = 2, alpha = 0.25)
```

It also supports grouping variables. Let's say we want to compare resistance of drugs against Urine Tract Infections (UTI) between hospitals A to D (variable `hospital_id`):
```r
septic_patients %>%
select(hospital_id, amox, nitr, fosf, trim, cipr) %>%
group_by(hospital_id) %>%
ggplot_rsi(x = "hospital_id",
facet = "Antibiotic",
nrow = 1,
datalabels = FALSE) +
labs(title = "AMR of Anti-UTI Drugs Per Hospital",
x = "Hospital")
```

You could use this to group on anything in your plots: Gram stain, age (group), genus, geographic location, et cetera.
#### MIC
```r
# Transform values to new class
mic_data <- as.mic(c(">=32", "1.0", "8", "<=0.128", "8", "16", "16"))
summary(mic_data)
# Mode:mic
# :0
# Min.:<=0.128
# Max.:>=32
plot(mic_data)
```

### Overwrite/force resistance based on EUCAST rules
This is also called *interpretive reading*.
```r
before <- data.frame(bact = c("STAAUR", # Staphylococcus aureus
"ENCFAE", # Enterococcus faecalis
"ESCCOL", # Escherichia coli
"KLEPNE", # Klebsiella pneumoniae
"PSEAER"), # Pseudomonas aeruginosa
vanc = "-", # Vancomycin
amox = "-", # Amoxicillin
coli = "-", # Colistin
cfta = "-", # Ceftazidime
cfur = "-", # Cefuroxime
stringsAsFactors = FALSE)
before
# bact vanc amox coli cfta cfur
# 1 STAAUR - - - - -
# 2 ENCFAE - - - - -
# 3 ESCCOL - - - - -
# 4 KLEPNE - - - - -
# 5 PSEAER - - - - -
# Now apply those rules; just need a column with bacteria IDs and antibiotic results:
after <- EUCAST_rules(before, col_mo = "bact")
after
# bact vanc amox coli cfta cfur
# 1 STAAUR - - R R -
# 2 ENCFAE - - R R R
# 3 ESCCOL R - - - -
# 4 KLEPNE R R - - -
# 5 PSEAER R R - - R
```
Bacteria IDs can be retrieved with the `guess_mo` function. It uses any type of info about a microorganism as input. For example, all these will return value `STAAUR`, the ID of *S. aureus*:
```r
guess_mo("stau")
guess_mo("STAU")
guess_mo("staaur")
guess_mo("S. aureus")
guess_mo("S aureus")
guess_mo("Staphylococcus aureus")
guess_mo("MRSA") # Methicillin Resistant S. aureus
guess_mo("VISA") # Vancomycin Intermediate S. aureus
guess_mo("VRSA") # Vancomycin Resistant S. aureus
```
### Other (microbial) epidemiological functions
```r
# G-test to replace Chi squared test
g.test(...)
# Determine key antibiotic based on bacteria ID
key_antibiotics(...)
# Selection of first isolates of any patient
first_isolate(...)
# Calculate resistance levels of antibiotics, can be used with `summarise` (dplyr)
rsi(...)
# Predict resistance levels of antibiotics
rsi_predict(...)
# Get name of antibiotic by ATC code
abname(...)
abname("J01CR02", from = "atc", to = "umcg") # "AMCL"
```
### Frequency tables
Base R lacks a simple function to create frequency tables. We created such a function that works with almost all data types: `freq` (or `frequency_tbl`). It can be used in two ways:
```r
# Like base R:
freq(mydata$myvariable)
# And like tidyverse:
mydata %>% freq(myvariable)
```
Factors sort on item by default:
```r
septic_patients %>% freq(hospital_id)
# Frequency table of `hospital_id`
# Class: factor
# Length: 2000 (of which NA: 0 = 0.0%)
# Unique: 4
#
# Item Count Percent Cum. Count Cum. Percent (Factor Level)
# --- ----- ------ -------- ----------- ------------- ---------------
# 1 A 319 16.0% 319 16.0% 1
# 2 B 661 33.1% 980 49.0% 2
# 3 C 256 12.8% 1236 61.8% 3
# 4 D 764 38.2% 2000 100.0% 4
```
This can be changed with the `sort.count` parameter:
```r
septic_patients %>% freq(hospital_id, sort.count = TRUE)
# Frequency table of `hospital_id`
# Class: factor
# Length: 2000 (of which NA: 0 = 0.0%)
# Unique: 4
#
# Item Count Percent Cum. Count Cum. Percent (Factor Level)
# --- ----- ------ -------- ----------- ------------- ---------------
# 1 D 764 38.2% 764 38.2% 4
# 2 B 661 33.1% 1425 71.2% 2
# 3 A 319 16.0% 1744 87.2% 1
# 4 C 256 12.8% 2000 100.0% 3
```
All other types, like numbers, characters and dates, sort on count by default:
```r
septic_patients %>% freq(date)
# Frequency table of `date`
# Class: Date
# Length: 2000 (of which NA: 0 = 0.0%)
# Unique: 1151
#
# Oldest: 2 January 2002
# Newest: 28 December 2017 (+5839)
# Median: 7 Augustus 2009 (~48%)
#
# Item Count Percent Cum. Count Cum. Percent
# --- ----------- ------ -------- ----------- -------------
# 1 2016-05-21 10 0.5% 10 0.5%
# 2 2004-11-15 8 0.4% 18 0.9%
# 3 2013-07-29 8 0.4% 26 1.3%
# 4 2017-06-12 8 0.4% 34 1.7%
# 5 2015-11-19 7 0.4% 41 2.1%
# 6 2005-12-22 6 0.3% 47 2.4%
# 7 2015-10-12 6 0.3% 53 2.6%
# 8 2002-05-16 5 0.2% 58 2.9%
# 9 2004-02-02 5 0.2% 63 3.1%
# 10 2004-02-18 5 0.2% 68 3.4%
# 11 2005-08-16 5 0.2% 73 3.6%
# 12 2005-09-01 5 0.2% 78 3.9%
# 13 2006-06-29 5 0.2% 83 4.2%
# 14 2007-08-10 5 0.2% 88 4.4%
# 15 2008-08-29 5 0.2% 93 4.7%
# [ reached getOption("max.print.freq") -- omitted 1136 entries, n = 1907 (95.3%) ]
```
For numeric values, some extra descriptive statistics will be calculated:
```r
freq(runif(n = 10, min = 1, max = 5))
# Frequency table
# Class: numeric
# Length: 10 (of which NA: 0 = 0.0%)
# Unique: 10
#
# Mean: 3.4
# Std. dev.: 1.3 (CV: 0.38, MAD: 1.3)
# Five-Num: 1.6 | 2.0 | 3.9 | 4.7 | 4.8 (IQR: 2.7, CQV: 0.4)
# Outliers: 0
#
# Item Count Percent Cum. Count Cum. Percent
# --- --------- ------ -------- ----------- -------------
# 1 1.568997 1 10.0% 1 10.0%
# 2 1.993575 1 10.0% 2 20.0%
# 3 2.022348 1 10.0% 3 30.0%
# 4 2.236038 1 10.0% 4 40.0%
# 5 3.579828 1 10.0% 5 50.0%
# 6 4.178081 1 10.0% 6 60.0%
# 7 4.394818 1 10.0% 7 70.0%
# 8 4.689871 1 10.0% 8 80.0%
# 9 4.698626 1 10.0% 9 90.0%
# 10 4.751488 1 10.0% 10 100.0%
#
# Warning message:
# All observations are unique.
```
Learn more about this function with:
```r
?freq
```
### Data sets included in package
Data sets to work with antibiotics and bacteria properties.
```r
# Data set with complete taxonomic trees from ITIS, containing of
# the three kingdoms Bacteria, Fungi and Protozoa
microorganisms # A tibble: 18,831 x 15
# Data set with 2000 random blood culture isolates from anonymised
# septic patients between 2001 and 2017 in 5 Dutch hospitals
septic_patients # A tibble: 2,000 x 49
# Data set with ATC antibiotics codes, official names, trade names
# and DDDs (oral and parenteral)
antibiotics # A tibble: 423 x 18
```
## Benchmarks
One of the most important features of this package is the complete microbial taxonomic database, supplied by ITIS (https://www.itis.gov). We created a function `as.mo` that transforms any user input value to a valid microbial ID by using AI (Artificial Intelligence) and based on the taxonomic tree of ITIS.
Using the `microbenchmark` package, we can review the calculation performance of this function.
```r
library(microbenchmark)
```
In the next test, we try to 'coerce' different input values for *Staphylococcus aureus*. The actual result is the same every time: it returns its MO code `B_STAPHY_AUR` (*B* stands for *Bacteria*, the taxonomic kingdom).
But the calculation time differs a lot. Here, the AI effect can be reviewed best:
```r
microbenchmark(A = as.mo("stau"),
B = as.mo("staaur"),
C = as.mo("S. aureus"),
D = as.mo("S. aureus"),
E = as.mo("STAAUR"),
F = as.mo("Staphylococcus aureus"),
times = 10,
unit = "ms")
# Unit: milliseconds
# expr min lq mean median uq max neval
# A 36.05088 36.14782 36.65635 36.24466 36.43075 39.78544 10
# B 16.43575 16.46885 16.67816 16.66053 16.84858 16.95299 10
# C 14.44150 14.52182 16.81197 14.59173 14.67854 36.75244 10
# D 14.49765 14.58153 16.71666 14.59414 14.61094 35.50731 10
# E 14.45212 14.75146 14.82033 14.85559 14.96433 15.03438 10
# F 10.69445 10.73852 10.80334 10.79596 10.86856 10.97465 10
```
The more an input value resembles a full name, the faster the result will be found. In the table above, all measurements are in milliseconds, tested on a quite regular Linux server from 2007 with 2 GB RAM. A value of 10.8 milliseconds means it can roughly determine 93 different input values per second. It case of 36.2 milliseconds, this is only 28 input values per second.
To improve speed, the `as.mo` function also takes into account the prevalence of human pathogenic microorganisms. The downside is of course that less prevalent microorganisms will be determined far less faster. See this example for the ID of *Burkholderia nodosa* (`B_BRKHL_NOD`):
```r
microbenchmark(B = as.mo("burnod"),
C = as.mo("B. nodosa"),
D = as.mo("B. nodosa"),
E = as.mo("BURNOD"),
F = as.mo("Burkholderia nodosa"),
times = 10,
unit = "ms")
# Unit: milliseconds
# expr min lq mean median uq max neval
# B 175.9446 176.80440 179.18240 177.00131 177.62021 198.86286 10
# C 88.1902 88.57705 89.08851 88.84293 89.15498 91.76621 10
# D 110.2641 110.67497 113.66290 111.20534 111.80744 134.44699 10
# E 175.0728 177.04235 207.83542 190.38109 200.33448 388.12177 10
# F 45.0778 45.31617 52.72430 45.62962 67.85262 70.42250 10
```
(Note: `A` is missing here, because `as.mo("buno")` returns `F_BUELL_NOT`: the ID of the fungus *Buellia notabilis*)
That takes up to 12 times as much time! A value of 190.4 milliseconds means it can only determine 5 different input values per second. We can conclude that looking up arbitrary codes of less prevalent microorganisms is the worst way to go, in terms of calculation performance.
To relieve this pitfall and further improve performance, two important calculations take almost no time at all: **repetive results** and **already precalculated results**.
Let's set up 25,000 entries of `"Staphylococcus aureus"` and check its speed:
```r
repetive_results <- rep("Staphylococcus aureus", 25000)
microbenchmark(A = as.mo(repetive_results),
times = 10,
unit = "ms")
# Unit: milliseconds
# expr min lq mean median uq max neval
# A 14.61282 14.6372 14.70817 14.72597 14.76124 14.78498 10
```
So transforming 25,000 times (!) `"Staphylococcus aureus"` only takes 4 ms (0.004 seconds) more than transforming it once. You only lose time on your unique input values.
What about precalculated results? This package also contains helper functions for specific microbial properties, for example `mo_fullname`. It returns the full microbial name (genus, species and possibly subspecies) and uses `as.mo` internally. If the input is however an already precalculated result, it almost doesn't take any time at all (see 'C' below):
```r
microbenchmark(A = mo_fullname("B_STPHY_AUR"),
B = mo_fullname("S. aureus"),
C = mo_fullname("Staphylococcus aureus"),
times = 10,
unit = "ms")
# Unit: milliseconds
# expr min lq mean median uq max neval
# A 13.548652 13.74588 13.8052969 13.813594 13.881165 14.090969 10
# B 15.079781 15.16785 15.3835842 15.374477 15.395115 16.072995 10
# C 0.171182 0.18563 0.2306307 0.203413 0.224610 0.492312 10
```
So going from `mo_fullname("Staphylococcus aureus")` to `"Staphylococcus aureus"` takes 0.0002 seconds - it doesn't even start calculating *if the result would be the same as the expected resulting value*. That goes for all helper functions:
```r
microbenchmark(A = mo_species("aureus"),
B = mo_genus("Staphylococcus"),
C = mo_fullname("Staphylococcus aureus"),
D = mo_family("Staphylococcaceae"),
E = mo_order("Bacillales"),
F = mo_class("Bacilli"),
G = mo_phylum("Firmicutes"),
H = mo_subkingdom("Posibacteria"),
times = 10,
unit = "ms")
# Unit: milliseconds
# expr min lq mean median uq max neval
# A 0.145270 0.158750 0.1908419 0.1693655 0.218255 0.300528 10
# B 0.182985 0.184522 0.2025408 0.1970235 0.209944 0.243328 10
# C 0.176280 0.201632 0.2618147 0.2303025 0.339499 0.388249 10
# D 0.136890 0.139054 0.1552231 0.1518010 0.168738 0.193042 10
# E 0.100921 0.116496 0.1321823 0.1222930 0.129976 0.230477 10
# F 0.103017 0.110281 0.1214480 0.1199880 0.124319 0.147506 10
# G 0.099246 0.110280 0.1195553 0.1188705 0.125436 0.149741 10
# H 0.114331 0.117264 0.1249819 0.1220830 0.129557 0.143385 10
```
Of course, when running `mo_phylum("Firmicutes")` the function has zero knowledge about the actual microorganism, namely *S. aureus*. But since the result would be `"Firmicutes"` too, there is no point in calculating the result. And since this package 'knows' all phyla of all known microorganisms (according to ITIS), it can just return the initial value immediately.
## Copyright
[](https://github.com/msberends/AMR/blob/master/LICENSE)
This R package is licensed under the [GNU General Public License (GPL) v2.0](https://github.com/msberends/AMR/blob/master/LICENSE). In a nutshell, this means that this package:
- May be used for commercial purposes
- May be used for private purposes
- May **not** be used for patent purposes
- May be modified, although:
- Modifications **must** be released under the same license when distributing the package
- Changes made to the code **must** be documented
- May be distributed, although:
- Source code **must** be made available when the package is distributed
- A copy of the license and copyright notice **must** be included with the package.
- Comes with a LIMITATION of liability
- Comes with NO warranty