This algorithm is used by as.mo() and all the mo_* functions to determine the most probable match of taxonomic records based on user input.
Arguments
- x
Any user input value(s)
- n
A full taxonomic name, that exists in
microorganisms$fullname
Note
This algorithm was originally described in: Berends MS et al. (2022). AMR: An R Package for Working with Antimicrobial Resistance Data. Journal of Statistical Software, 104(3), 1-31; doi:10.18637/jss.v104.i03 .
Later, the work of Bartlett A et al. about bacterial pathogens infecting humans (2022, doi:10.1099/mic.0.001269 ) was incorporated.
Matching Score for Microorganisms
With ambiguous user input in as.mo() and all the mo_* functions, the returned results are chosen based on their matching score using mo_matching_score(). This matching score \(m\), is calculated as:
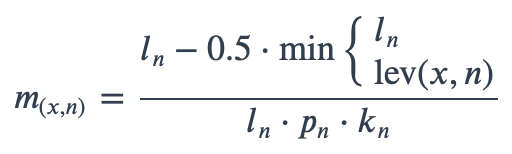
where:
\(x\) is the user input;
\(n\) is a taxonomic name (genus, species, and subspecies);
\(l_n\) is the length of \(n\);
\(lev\) is the Levenshtein distance function (counting any insertion as 1, and any deletion or substitution as 2) that is needed to change \(x\) into \(n\);
\(p_n\) is the human pathogenic prevalence group of \(n\), as described below;
\(k_n\) is the taxonomic kingdom of \(n\), set as Bacteria = 1, Fungi = 1.25, Protozoa = 1.5, Archaea = 2, others = 3.
The grouping into human pathogenic prevalence \(p\) is based on recent work from Bartlett et al. (2022, doi:10.1099/mic.0.001269 ) who extensively studied medical-scientific literature to categorise all bacterial species into these groups:
Established, if a taxonomic species has infected at least three persons in three or more references. These records have
prevalence = 1.0in the microorganisms data set;Putative, if a taxonomic species has fewer than three known cases. These records have
prevalence = 1.25in the microorganisms data set.
Furthermore,
Any genus present in the established list also has
prevalence = 1.0in the microorganisms data set;Any other genus present in the putative list has
prevalence = 1.25in the microorganisms data set;Any other species or subspecies of which the genus is present in the two aforementioned groups, has
prevalence = 1.5in the microorganisms data set;Any non-bacterial genus, species or subspecies of which the genus is present in the following list, has
prevalence = 1.25in the microorganisms data set: Absidia, Acanthamoeba, Acremonium, Aedes, Alternaria, Amoeba, Ancylostoma, Angiostrongylus, Anisakis, Anopheles, Apophysomyces, Arthroderma, Aspergillus, Aureobasidium, Basidiobolus, Beauveria, Blastocystis, Blastomyces, Candida, Capillaria, Chaetomium, Chrysonilia, Chrysosporium, Cladophialophora, Cladosporium, Conidiobolus, Contracaecum, Cordylobia, Cryptococcus, Curvularia, Demodex, Dermatobia, Dientamoeba, Diphyllobothrium, Dirofilaria, Echinostoma, Entamoeba, Enterobius, Exophiala, Exserohilum, Fasciola, Fonsecaea, Fusarium, Geotrichum, Giardia, Haloarcula, Halobacterium, Halococcus, Hendersonula, Heterophyes, Histomonas, Histoplasma, Hymenolepis, Hypomyces, Hysterothylacium, Kloeckera, Kodamaea, Leishmania, Lichtheimia, Lodderomyces, Malassezia, Malbranchea, Metagonimus, Meyerozyma, Microsporidium, Microsporum, Millerozyma, Mortierella, Mucor, Mycocentrospora, Necator, Nectria, Ochroconis, Oesophagostomum, Oidiodendron, Opisthorchis, Paecilomyces, Pediculus, Penicillium, Phlebotomus, Phoma, Pichia, Piedraia, Pithomyces, Pityrosporum, Pneumocystis, Pseudallescheria, Pseudoterranova, Pulex, Rhizomucor, Rhizopus, Rhodotorula, Saccharomyces, Saprochaete, Sarcoptes, Scedosporium, Scolecobasidium, Scopulariopsis, Scytalidium, Spirometra, Sporobolomyces, Sporotrichum, Stachybotrys, Strongyloides, Syngamus, Taenia, Talaromyces, Toxocara, Trichinella, Trichobilharzia, Trichoderma, Trichomonas, Trichophyton, Trichosporon, Trichostrongylus, Trichuris, Tritirachium, Trombicula, Trypanosoma, Tunga, Verticillium, or Wuchereria;All other records have
prevalence = 2.0in the microorganisms data set.
When calculating the matching score, all characters in \(x\) and \(n\) are ignored that are other than A-Z, a-z, 0-9, spaces and parentheses.
All matches are sorted descending on their matching score and for all user input values, the top match will be returned. This will lead to the effect that e.g., "E. coli" will return the microbial ID of Escherichia coli (\(m = 0.688\), a highly prevalent microorganism found in humans) and not Entamoeba coli (\(m = 0.381\), a less prevalent microorganism in humans), although the latter would alphabetically come first.
Reference Data Publicly Available
All data sets in this AMR package (about microorganisms, antibiotics, SIR interpretation, EUCAST rules, etc.) are publicly and freely available for download in the following formats: R, MS Excel, Apache Feather, Apache Parquet, SPSS, SAS, and Stata. We also provide tab-separated plain text files that are machine-readable and suitable for input in any software program, such as laboratory information systems. Please visit our website for the download links. The actual files are of course available on our GitHub repository.
Examples
mo_reset_session()
#> ℹ Reset 17 previously matched input values.
as.mo("E. coli")
#> Class 'mo'
#> [1] B_ESCHR_COLI
mo_uncertainties()
#> Matching scores are based on the resemblance between the input and the full
#> taxonomic name, and the pathogenicity in humans. See ?mo_matching_score.
#>
#> --------------------------------------------------------------------------------
#> "E. coli" -> Escherichia coli (B_ESCHR_COLI, 0.688)
#> Also matched: Enterobacter cowanii (0.600), Eubacterium combesii
#> (0.600), Eggerthia catenaformis (0.591), Eubacterium callanderi
#> (0.591), Enterocloster citroniae (0.587), Eubacterium cylindroides
#> (0.583), Enterococcus casseliflavus (0.577), Enterobacter cloacae
#> cloacae (0.571), Enterobacter cloacae complex (0.571), and Ehrlichia
#> canis (0.567)
#>
#> Only the first 10 other matches of each record are shown. Run
#> print(mo_uncertainties(), n = ...) to view more entries, or save
#> mo_uncertainties() to an object.
mo_matching_score(
x = "E. coli",
n = c("Escherichia coli", "Entamoeba coli")
)
#> [1] 0.6875000 0.3809524