Clean up existing SIR values, or interpret minimum inhibitory concentration (MIC) values and disk diffusion diameters according to EUCAST or CLSI. as.sir() transforms the input to a new class sir, which is an ordered factor containing the levels S, SDD, I, R, NI.
Breakpoints are currently implemented from EUCAST 2011-2025 and CLSI 2011-2025, see Details. All breakpoints used for interpretation are available in our clinical_breakpoints data set.
Usage
as.sir(x, ...)
NA_sir_
is.sir(x)
is_sir_eligible(x, threshold = 0.05)
# Default S3 method
as.sir(x, S = "^(S|U)+$", I = "^(I)+$", R = "^(R)+$",
NI = "^(N|NI|V)+$", SDD = "^(SDD|D|H)+$", info = interactive(), ...)
# S3 method for class 'mic'
as.sir(x, mo = NULL, ab = deparse(substitute(x)),
guideline = getOption("AMR_guideline", "EUCAST"), uti = NULL,
capped_mic_handling = getOption("AMR_capped_mic_handling", "standard"),
add_intrinsic_resistance = FALSE,
reference_data = AMR::clinical_breakpoints,
substitute_missing_r_breakpoint = getOption("AMR_substitute_missing_r_breakpoint",
FALSE), include_screening = getOption("AMR_include_screening", FALSE),
include_PKPD = getOption("AMR_include_PKPD", TRUE),
breakpoint_type = getOption("AMR_breakpoint_type", "human"), host = NULL,
language = get_AMR_locale(), verbose = FALSE, info = interactive(),
conserve_capped_values = NULL, ...)
# S3 method for class 'disk'
as.sir(x, mo = NULL, ab = deparse(substitute(x)),
guideline = getOption("AMR_guideline", "EUCAST"), uti = NULL,
add_intrinsic_resistance = FALSE,
reference_data = AMR::clinical_breakpoints,
substitute_missing_r_breakpoint = getOption("AMR_substitute_missing_r_breakpoint",
FALSE), include_screening = getOption("AMR_include_screening", FALSE),
include_PKPD = getOption("AMR_include_PKPD", TRUE),
breakpoint_type = getOption("AMR_breakpoint_type", "human"), host = NULL,
language = get_AMR_locale(), verbose = FALSE, info = interactive(),
...)
# S3 method for class 'data.frame'
as.sir(x, ..., col_mo = NULL,
guideline = getOption("AMR_guideline", "EUCAST"), uti = NULL,
capped_mic_handling = getOption("AMR_capped_mic_handling", "standard"),
add_intrinsic_resistance = FALSE,
reference_data = AMR::clinical_breakpoints,
substitute_missing_r_breakpoint = getOption("AMR_substitute_missing_r_breakpoint",
FALSE), include_screening = getOption("AMR_include_screening", FALSE),
include_PKPD = getOption("AMR_include_PKPD", TRUE),
breakpoint_type = getOption("AMR_breakpoint_type", "human"), host = NULL,
language = get_AMR_locale(), verbose = FALSE, info = interactive(),
parallel = FALSE, max_cores = -1, conserve_capped_values = NULL)
sir_interpretation_history(clean = FALSE)Source
For interpretations of minimum inhibitory concentration (MIC) values and disk diffusion diameters:
CLSI M39: Analysis and Presentation of Cumulative Antimicrobial Susceptibility Test Data, 2011-2025, Clinical and Laboratory Standards Institute (CLSI). https://clsi.org/standards/products/microbiology/documents/m39/.
CLSI M100: Performance Standard for Antimicrobial Susceptibility Testing, 2011-2025, Clinical and Laboratory Standards Institute (CLSI). https://clsi.org/standards/products/microbiology/documents/m100/.
CLSI VET01: Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated From Animals, 2019-2025, Clinical and Laboratory Standards Institute (CLSI). https://clsi.org/standards/products/veterinary-medicine/documents/vet01/.
EUCAST Breakpoint tables for interpretation of MICs and zone diameters, 2011-2025, European Committee on Antimicrobial Susceptibility Testing (EUCAST). https://www.eucast.org/clinical_breakpoints.
WHONET as a source for machine-reading the clinical breakpoints (read more here), 1989-2025, WHO Collaborating Centre for Surveillance of Antimicrobial Resistance. https://whonet.org/.
Arguments
- x
Vector of values (for class
mic: MIC values in mg/L, for classdisk: a disk diffusion radius in millimetres).- ...
For using on a data.frame: selection of columns to apply
as.sir()to. Supports tidyselect language such aswhere(is.mic),starts_with(...), orcolumn1:column4, and can thus also be antimicrobial selectors such asas.sir(df, penicillins()).Otherwise: arguments passed on to methods.
- threshold
Maximum fraction of invalid antimicrobial interpretations of
x, see Examples.- S, I, R, NI, SDD
A case-independent regular expression to translate input to this result. This regular expression will be run after all non-letters and whitespaces are removed from the input.
- info
A logical to print information about the process, defaults to
TRUEonly in interactive sessions.- mo
A vector (or column name) with characters that can be coerced to valid microorganism codes with
as.mo(), can be left empty to determine it automatically.- ab
A vector (or column name) with characters that can be coerced to a valid antimicrobial drug code with
as.ab().- guideline
A guideline name (or column name) to use for SIR interpretation. Defaults to EUCAST 2025 (the latest implemented EUCAST guideline in the clinical_breakpoints data set), but can be set with the package option
AMR_guideline. Currently supports EUCAST (2011-2025) and CLSI (2011-2025), see Details. Using a column name allows for straightforward interpretation of historical data, which must be analysed in the context of, for example, different years.- uti
(Urinary Tract Infection) a vector (or column name) with logicals (
TRUEorFALSE) to specify whether a UTI specific interpretation from the guideline should be chosen. For usingas.sir()on a data.frame, this can also be a column containing logicals or when left blank, the data set will be searched for a column 'specimen', and rows within this column containing 'urin' (such as 'urine', 'urina') will be regarded isolates from a UTI. See Examples.- capped_mic_handling
A character string that controls how MIC values with a cap (i.e., starting with
<,<=,>, or>=) are interpreted. Supports the following options:"none"<=and>=are treated as-is.<and>are treated as-is.
"conservative"<=and>=return"NI"(non-interpretable) if the MIC is within the breakpoint guideline range.<always returns"S", and>always returns"R".
"standard"(default)<=and>=return"NI"(non-interpretable) if the MIC is within the breakpoint guideline range.<and>are treated as-is.
"inverse"<=and>=are treated as-is.<always returns"S", and>always returns"R".
The default
"standard"setting ensures cautious handling of uncertain values while preserving interpretability. This option can also be set with the package optionAMR_capped_mic_handling.- add_intrinsic_resistance
(only useful when using a EUCAST guideline) a logical to indicate whether intrinsic antibiotic resistance must also be considered for applicable bug-drug combinations, meaning that e.g. ampicillin will always return "R" in Klebsiella species. Determination is based on the intrinsic_resistant data set, that itself is based on 'EUCAST Expert Rules' and 'EUCAST Intrinsic Resistance and Unusual Phenotypes' v3.3 (2021).
- reference_data
A data.frame to be used for interpretation, which defaults to the clinical_breakpoints data set. Changing this argument allows for using own interpretation guidelines. This argument must contain a data set that is equal in structure to the clinical_breakpoints data set (same column names and column types). Please note that the
guidelineargument will be ignored whenreference_datais manually set.- substitute_missing_r_breakpoint
A logical to indicate that a missing clinical breakpoints for R (resistant) must be substituted with R - the default is
FALSE. Some (especially CLSI) breakpoints only have a breakpoint for S, meaning that the outcome can only be"S"orNA. Setting this toTRUEwill convert theNAs in these cases to"R". Can also be set with the package optionAMR_substitute_missing_r_breakpoint.- include_screening
A logical to indicate that clinical breakpoints for screening are allowed - the default is
FALSE. Can also be set with the package optionAMR_include_screening.- include_PKPD
A logical to indicate that PK/PD clinical breakpoints must be applied as a last resort - the default is
TRUE. Can also be set with the package optionAMR_include_PKPD.- breakpoint_type
The type of breakpoints to use, either "ECOFF", "animal", or "human". ECOFF stands for Epidemiological Cut-Off values. The default is
"human", which can also be set with the package optionAMR_breakpoint_type. Ifhostis set to values of veterinary species, this will automatically be set to"animal".- host
A vector (or column name) with characters to indicate the host. Only useful for veterinary breakpoints, as it requires
breakpoint_type = "animal". The values can be any text resembling the animal species, even in any of the 28 supported languages of this package. For foreign languages, be sure to set the language withset_AMR_locale()(though it will be automatically guessed based on the system language).- language
Language to convert values set in
hostwhen using animal breakpoints. Use one of these supported language names or ISO 639-1 codes: English (en), Arabic (ar), Bengali (bn), Chinese (zh), Czech (cs), Danish (da), Dutch (nl), Finnish (fi), French (fr), German (de), Greek (el), Hindi (hi), Indonesian (id), Italian (it), Japanese (ja), Korean (ko), Norwegian (no), Polish (pl), Portuguese (pt), Romanian (ro), Russian (ru), Spanish (es), Swahili (sw), Swedish (sv), Turkish (tr), Ukrainian (uk), Urdu (ur), or Vietnamese (vi).- verbose
A logical to indicate that all notes should be printed during interpretation of MIC values or disk diffusion values.
- conserve_capped_values
Deprecated, use
capped_mic_handlinginstead.- col_mo
Column name of the names or codes of the microorganisms (see
as.mo()) - the default is the first column of classmo. Values will be coerced usingas.mo().- parallel
A logical to indicate if parallel computing must be used, defaults to
FALSE. This requires no additional packages, as the usedparallelpackage is part of base R. On Windows and on R < 4.0.0parallel::parLapply()will be used, in all other cases the more efficientparallel::mclapply()will be used.- max_cores
Maximum number of cores to use if
parallel = TRUE. Use a negative value to subtract that number from the available number of cores, e.g. a value of-2on an 8-core machine means that at most 6 cores will be used. Defaults to-1. There will never be used more cores than variables to analyse. The available number of cores are detected usingparallelly::availableCores()if that package is installed, and base R'sparallel::detectCores()otherwise.- clean
A logical to indicate whether previously stored results should be forgotten after returning the 'logbook' with results.
Value
Ordered factor with new class sir
Details
Note: The clinical breakpoints in this package were validated through, and imported from, WHONET. The public use of this AMR package has been endorsed by both CLSI and EUCAST. See clinical_breakpoints for more information.
How it Works
The as.sir() function can work in four ways:
For cleaning raw / untransformed data. The data will be cleaned to only contain valid values, namely: S for susceptible, I for intermediate or 'susceptible, increased exposure', R for resistant, NI for non-interpretable, and SDD for susceptible dose-dependent. Each of these can be set using a regular expression. Furthermore,
as.sir()will try its best to clean with some intelligence. For example, mixed values with SIR interpretations and MIC values such as"<0.25; S"will be coerced to"S". Combined interpretations for multiple test methods (as seen in laboratory records) such as"S; S"will be coerced to"S", but a value like"S; I"will returnNAwith a warning that the input is invalid.For interpreting minimum inhibitory concentration (MIC) values according to EUCAST or CLSI. You must clean your MIC values first using
as.mic(), that also gives your columns the new data classmic. Also, be sure to have a column with microorganism names or codes. It will be found automatically, but can be set manually using themoargument.Example to apply using
dplyr:your_data %>% mutate_if(is.mic, as.sir) your_data %>% mutate(across(where(is.mic), as.sir)) your_data %>% mutate_if(is.mic, as.sir, ab = "column_with_antibiotics", mo = "column_with_microorganisms") your_data %>% mutate_if(is.mic, as.sir, ab = c("cipro", "ampicillin", ...), mo = c("E. coli", "K. pneumoniae", ...)) # for veterinary breakpoints, also set `host`: your_data %>% mutate_if(is.mic, as.sir, host = "column_with_animal_species", guideline = "CLSI") # fast processing with parallel computing: as.sir(your_data, ..., parallel = TRUE)Operators like "<=" will be stripped before interpretation. When using
capped_mic_handling = "conservative", an MIC value of e.g. ">2" will always return "R", even if the breakpoint according to the chosen guideline is ">=4". This is to prevent that capped values from raw laboratory data would not be treated conservatively. The default behaviour (capped_mic_handling = "standard") considers ">2" to be lower than ">=4" and might in this case return "S" or "I".Note: When using CLSI as the guideline, MIC values must be log2-based doubling dilutions. Values not in this format, will be automatically rounded up to the nearest log2 level as CLSI instructs, and a warning will be thrown.
For interpreting disk diffusion diameters according to EUCAST or CLSI. You must clean your disk zones first using
as.disk(), that also gives your columns the new data classdisk. Also, be sure to have a column with microorganism names or codes. It will be found automatically, but can be set manually using themoargument.Example to apply using
dplyr:your_data %>% mutate_if(is.disk, as.sir) your_data %>% mutate(across(where(is.disk), as.sir)) your_data %>% mutate_if(is.disk, as.sir, ab = "column_with_antibiotics", mo = "column_with_microorganisms") your_data %>% mutate_if(is.disk, as.sir, ab = c("cipro", "ampicillin", ...), mo = c("E. coli", "K. pneumoniae", ...)) # for veterinary breakpoints, also set `host`: your_data %>% mutate_if(is.disk, as.sir, host = "column_with_animal_species", guideline = "CLSI") # fast processing with parallel computing: as.sir(your_data, ..., parallel = TRUE)
For interpreting a complete data set, with automatic determination of MIC values, disk diffusion diameters, microorganism names or codes, and antimicrobial test results. This is done very simply by running
as.sir(your_data).
For points 2, 3 and 4: Use sir_interpretation_history() to retrieve a data.frame with all results of all previous as.sir() calls. It also contains notes about interpretation, and the exact input and output values.
Supported Guidelines
For interpreting MIC values as well as disk diffusion diameters, currently implemented guidelines are:
For clinical microbiology: EUCAST 2011-2025 and CLSI 2011-2025;
For veterinary microbiology: EUCAST 2021-2025 and CLSI 2019-2025;
For ECOFFs (Epidemiological Cut-off Values): EUCAST 2020-2025 and CLSI 2022-2025.
The guideline argument must be set to e.g., "EUCAST 2025" or "CLSI 2025". By simply using "EUCAST" (the default) or "CLSI" as input, the latest included version of that guideline will automatically be selected. Importantly, using a column name of your data instead, allows for straightforward interpretation of historical data that must be analysed in the context of, for example, different years.
You can set your own data set using the reference_data argument. The guideline argument will then be ignored.
It is also possible to set the default guideline with the package option AMR_guideline (e.g. in your .Rprofile file), such as:
Working with Veterinary Breakpoints
When using veterinary breakpoints (i.e., setting breakpoint_type = "animal"), a column with animal species must be available or set manually using the host argument. The column must contain names like "dogs", "cats", "cattle", "swine", "horses", "poultry", or "aquatic". Other animal names like "goats", "rabbits", or "monkeys" are also recognised but may not be available in all guidelines. Matching is case-insensitive and accepts Latin-based synonyms (e.g., "bovine" for cattle and "canine" for dogs).
Regarding choice of veterinary guidelines, these might be the best options to set before analysis:
After Interpretation
After using as.sir(), you can use the eucast_rules() defined by EUCAST to (1) apply inferred susceptibility and resistance based on results of other antimicrobials and (2) apply intrinsic resistance based on taxonomic properties of a microorganism.
To determine which isolates are multi-drug resistant, be sure to run mdro() (which applies the MDR/PDR/XDR guideline from 2012 at default) on a data set that contains S/I/R values. Read more about interpreting multidrug-resistant organisms here.
Other
The function is.sir() detects if the input contains class sir. If the input is a data.frame or list, it iterates over all columns/items and returns a logical vector.
The base R function as.double() can be used to retrieve quantitative values from a sir object: "S" = 1, "I"/"SDD" = 2, "R" = 3. All other values are rendered NA. Note: Do not use as.integer(), since that (because of how R works internally) will return the factor level indices, and not these aforementioned quantitative values.
The function is_sir_eligible() returns TRUE when a column contains at most 5% potentially invalid antimicrobial interpretations, and FALSE otherwise. The threshold of 5% can be set with the threshold argument. If the input is a data.frame, it iterates over all columns and returns a logical vector.
NA_sir_ is a missing value of the new sir class, analogous to e.g. base R's NA_character_.
Interpretation of SIR
In 2019, the European Committee on Antimicrobial Susceptibility Testing (EUCAST) has decided to change the definitions of susceptibility testing categories S, I, and R (https://www.eucast.org/newsiandr).
This AMR package follows insight; use susceptibility() (equal to proportion_SI()) to determine antimicrobial susceptibility and count_susceptible() (equal to count_SI()) to count susceptible isolates.
Download Our Reference Data
All reference data sets in the AMR package - including information on microorganisms, antimicrobials, and clinical breakpoints - are freely available for download in multiple formats: R, MS Excel, Apache Feather, Apache Parquet, SPSS, and Stata.
For maximum compatibility, we also provide machine-readable, tab-separated plain text files suitable for use in any software, including laboratory information systems.
Visit our website for direct download links, or explore the actual files in our GitHub repository.
Examples
example_isolates
#> # A tibble: 2,000 × 46
#> date patient age gender ward mo PEN OXA FLC AMX
#> <date> <chr> <dbl> <chr> <chr> <mo> <sir> <sir> <sir> <sir>
#> 1 2002-01-02 A77334 65 F Clinical B_ESCHR_COLI R NA NA NA
#> 2 2002-01-03 A77334 65 F Clinical B_ESCHR_COLI R NA NA NA
#> 3 2002-01-07 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 4 2002-01-07 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 5 2002-01-13 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 6 2002-01-13 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 7 2002-01-14 462729 78 M Clinical B_STPHY_AURS R NA S R
#> 8 2002-01-14 462729 78 M Clinical B_STPHY_AURS R NA S R
#> 9 2002-01-16 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 10 2002-01-17 858515 79 F ICU B_STPHY_EPDR R NA S NA
#> # ℹ 1,990 more rows
#> # ℹ 36 more variables: AMC <sir>, AMP <sir>, TZP <sir>, CZO <sir>, FEP <sir>,
#> # CXM <sir>, FOX <sir>, CTX <sir>, CAZ <sir>, CRO <sir>, GEN <sir>,
#> # TOB <sir>, AMK <sir>, KAN <sir>, TMP <sir>, SXT <sir>, NIT <sir>,
#> # FOS <sir>, LNZ <sir>, CIP <sir>, MFX <sir>, VAN <sir>, TEC <sir>,
#> # TCY <sir>, TGC <sir>, DOX <sir>, ERY <sir>, CLI <sir>, AZM <sir>,
#> # IPM <sir>, MEM <sir>, MTR <sir>, CHL <sir>, COL <sir>, MUP <sir>, …
summary(example_isolates[, 1:10]) # see all SIR results at a glance
#> date patient age gender
#> Min. :2002-01-02 Length:2000 Min. : 0.00 Length:2000
#> 1st Qu.:2005-07-31 Class :character 1st Qu.:63.00 Class :character
#> Median :2009-07-31 Mode :character Median :74.00 Mode :character
#> Mean :2009-11-20 Mean :70.69
#> 3rd Qu.:2014-05-30 3rd Qu.:82.00
#> Max. :2017-12-28 Max. :97.00
#> ward mo PEN
#> Length:2000 Class :mo Class:sir
#> Class :character <NA> :0 %S :25.6% (n=417)
#> Mode :character Unique:90 %SDD : 0.0% (n=0)
#> #1 :B_ESCHR_COLI %I : 0.7% (n=11)
#> #2 :B_STPHY_CONS %R :73.7% (n=1201)
#> #3 :B_STPHY_AURS %NI : 0.0% (n=0)
#> OXA FLC AMX
#> Class:sir Class:sir Class:sir
#> %S :68.8% (n=251) %S :70.5% (n=665) %S :40.2% (n=543)
#> %SDD : 0.0% (n=0) %SDD : 0.0% (n=0) %SDD : 0.0% (n=0)
#> %I : 0.0% (n=0) %I : 0.0% (n=0) %I : 0.2% (n=3)
#> %R :31.2% (n=114) %R :29.5% (n=278) %R :59.6% (n=804)
#> %NI : 0.0% (n=0) %NI : 0.0% (n=0) %NI : 0.0% (n=0)
# create some example data sets, with combined MIC values and disk zones
df_wide <- data.frame(
microorganism = "Escherichia coli",
amoxicillin = as.mic(8),
cipro = as.mic(0.256),
tobra = as.disk(16),
genta = as.disk(18),
ERY = "R"
)
df_long <- data.frame(
bacteria = rep("Escherichia coli", 4),
antibiotic = c("amoxicillin", "cipro", "tobra", "genta"),
mics = as.mic(c(0.01, 1, 4, 8)),
disks = as.disk(c(6, 10, 14, 18)),
guideline = c("EUCAST 2021", "EUCAST 2022", "EUCAST 2023", "EUCAST 2024")
)
# and clean previous SIR interpretation logs
x <- sir_interpretation_history(clean = TRUE)
# For INTERPRETING disk diffusion and MIC values -----------------------
# most basic application:
as.sir(df_wide)
#> microorganism amoxicillin cipro tobra genta ERY
#> 1 Escherichia coli S I S S R
# return a 'logbook' about the results:
sir_interpretation_history()
#> # A tibble: 4 × 18
#> datetime index method ab_given mo_given host_given input_given
#> <dttm> <int> <chr> <chr> <chr> <chr> <chr>
#> 1 2025-09-12 14:58:24 1 MIC amoxicillin Escherich… human 8
#> 2 2025-09-12 14:58:25 1 MIC cipro Escherich… human 0.256
#> 3 2025-09-12 14:58:25 1 DISK tobra Escherich… human 16
#> 4 2025-09-12 14:58:25 1 DISK genta Escherich… human 18
#> # ℹ 11 more variables: ab <ab>, mo <mo>, host <chr>, input <chr>,
#> # outcome <sir>, notes <chr>, guideline <chr>, ref_table <chr>, uti <lgl>,
#> # breakpoint_S_R <chr>, site <chr>
# \donttest{
# using parallel computing, which is available in base R:
as.sir(df_wide, parallel = TRUE, info = TRUE)
#> ℹ Returning previously coerced values for various antimicrobials. Run
#> `ab_reset_session()` to reset this. This note will be shown once per
#> session.
#>
#> Running in parallel mode using 3 out of 4 cores, on columns 'amoxicillin',
#> 'cipro', 'tobra', 'genta', and 'ERY'...
#> DONE
#>
#>
#> ℹ Run `sir_interpretation_history()` to retrieve a logbook with all details
#> of the breakpoint interpretations.
#> microorganism amoxicillin cipro tobra genta ERY
#> 1 Escherichia coli S I S S R
## Using dplyr -------------------------------------------------
if (require("dplyr")) {
# approaches that all work without additional arguments:
df_wide %>% mutate_if(is.mic, as.sir)
df_wide %>% mutate_if(function(x) is.mic(x) | is.disk(x), as.sir)
df_wide %>% mutate(across(where(is.mic), as.sir))
df_wide %>% mutate_at(vars(amoxicillin:tobra), as.sir)
df_wide %>% mutate(across(amoxicillin:tobra, as.sir))
df_wide %>% mutate(across(aminopenicillins(), as.sir))
# approaches that all work with additional arguments:
df_long %>%
# given a certain data type, e.g. MIC values
mutate_if(is.mic, as.sir,
mo = "bacteria",
ab = "antibiotic",
guideline = "guideline"
)
df_long %>%
mutate(across(
where(is.mic),
function(x) {
as.sir(x,
mo = "bacteria",
ab = "antibiotic",
guideline = "CLSI"
)
}
))
df_wide %>%
# given certain columns, e.g. from 'cipro' to 'genta'
mutate_at(vars(cipro:genta), as.sir,
mo = "bacteria",
guideline = "CLSI"
)
df_wide %>%
mutate(across(
cipro:genta,
function(x) {
as.sir(x,
mo = "bacteria",
guideline = "CLSI"
)
}
))
# for veterinary breakpoints, add 'host':
df_long$animal_species <- c("cats", "dogs", "horses", "cattle")
df_long %>%
# given a certain data type, e.g. MIC values
mutate_if(is.mic, as.sir,
mo = "bacteria",
ab = "antibiotic",
host = "animal_species",
guideline = "CLSI"
)
df_long %>%
mutate(across(
where(is.mic),
function(x) {
as.sir(x,
mo = "bacteria",
ab = "antibiotic",
host = "animal_species",
guideline = "CLSI"
)
}
))
df_wide %>%
mutate_at(vars(cipro:genta), as.sir,
mo = "bacteria",
ab = "antibiotic",
host = "animal_species",
guideline = "CLSI"
)
df_wide %>%
mutate(across(
cipro:genta,
function(x) {
as.sir(x,
mo = "bacteria",
host = "animal_species",
guideline = "CLSI"
)
}
))
# to include information about urinary tract infections (UTI)
data.frame(
mo = "E. coli",
nitrofuratoin = c("<= 2", 32),
from_the_bladder = c(TRUE, FALSE)
) %>%
as.sir(uti = "from_the_bladder")
data.frame(
mo = "E. coli",
nitrofuratoin = c("<= 2", 32),
specimen = c("urine", "blood")
) %>%
as.sir() # automatically determines urine isolates
df_wide %>%
mutate_at(vars(cipro:genta), as.sir, mo = "E. coli", uti = TRUE)
}
#> ℹ For `aminopenicillins()` using column 'amoxicillin'
#> Warning: There was 1 warning in `mutate()`.
#> ℹ In argument: `across(...)`.
#> Caused by warning:
#> ! Some MICs were converted to the nearest higher log2 level, following the
#> CLSI interpretation guideline.
#> Warning: There was 1 warning in `mutate()`.
#> ℹ In argument: `cipro = (function (x, ...) ...`.
#> Caused by warning:
#> ! Some MICs were converted to the nearest higher log2 level, following the
#> CLSI interpretation guideline.
#> Warning: There was 1 warning in `mutate()`.
#> ℹ In argument: `across(...)`.
#> Caused by warning:
#> ! Some MICs were converted to the nearest higher log2 level, following the
#> CLSI interpretation guideline.
#> Warning: There was 1 warning in `mutate()`.
#> ℹ In argument: `mics = (function (x, ...) ...`.
#> Caused by warning:
#> ! Some MICs were converted to the nearest higher log2 level, following the
#> CLSI interpretation guideline.
#> Warning: There was 1 warning in `mutate()`.
#> ℹ In argument: `across(...)`.
#> Caused by warning:
#> ! Some MICs were converted to the nearest higher log2 level, following the
#> CLSI interpretation guideline.
#> Interpreting MIC values: 'antibiotic' (ASP, acetylspiramycin), CLSI 2025...
#> Interpreting disk diffusion zones: 'antibiotic' (ASP, acetylspiramycin),
#> CLSI 2025...
#> Interpreting disk diffusion zones: 'antibiotic' (ASP, acetylspiramycin),
#> CLSI 2025...
#> Warning: There was 1 warning in `mutate()`.
#> ℹ In argument: `cipro = (function (x, ...) ...`.
#> Caused by warning:
#> ! Some MICs were converted to the nearest higher log2 level, following the
#> CLSI interpretation guideline.
#> Warning: There was 1 warning in `mutate()`.
#> ℹ In argument: `across(...)`.
#> Caused by warning:
#> ! Some MICs were converted to the nearest higher log2 level, following the
#> CLSI interpretation guideline.
#> microorganism amoxicillin cipro tobra genta ERY
#> 1 Escherichia coli 8 <NA> S S R
## Using base R ------------------------------------------------
# for single values
as.sir(
x = as.mic(2),
mo = as.mo("S. pneumoniae"),
ab = "AMP",
guideline = "EUCAST"
)
#> Class 'sir'
#> [1] R
as.sir(
x = as.disk(18),
mo = "Strep pneu", # `mo` will be coerced with as.mo()
ab = "ampicillin", # and `ab` with as.ab()
guideline = "EUCAST"
)
#> Class 'sir'
#> [1] R
# For CLEANING existing SIR values -------------------------------------
as.sir(c("S", "SDD", "I", "R", "NI", "A", "B", "C"))
#> Warning: in `as.sir()`: 3 results in index '20' truncated (38%) that were invalid
#> antimicrobial interpretations: "A", "B", and "C"
#> Class 'sir'
#> [1] S SDD I R NI <NA> <NA> <NA>
as.sir("<= 0.002; S") # will return "S"
#> Class 'sir'
#> [1] S
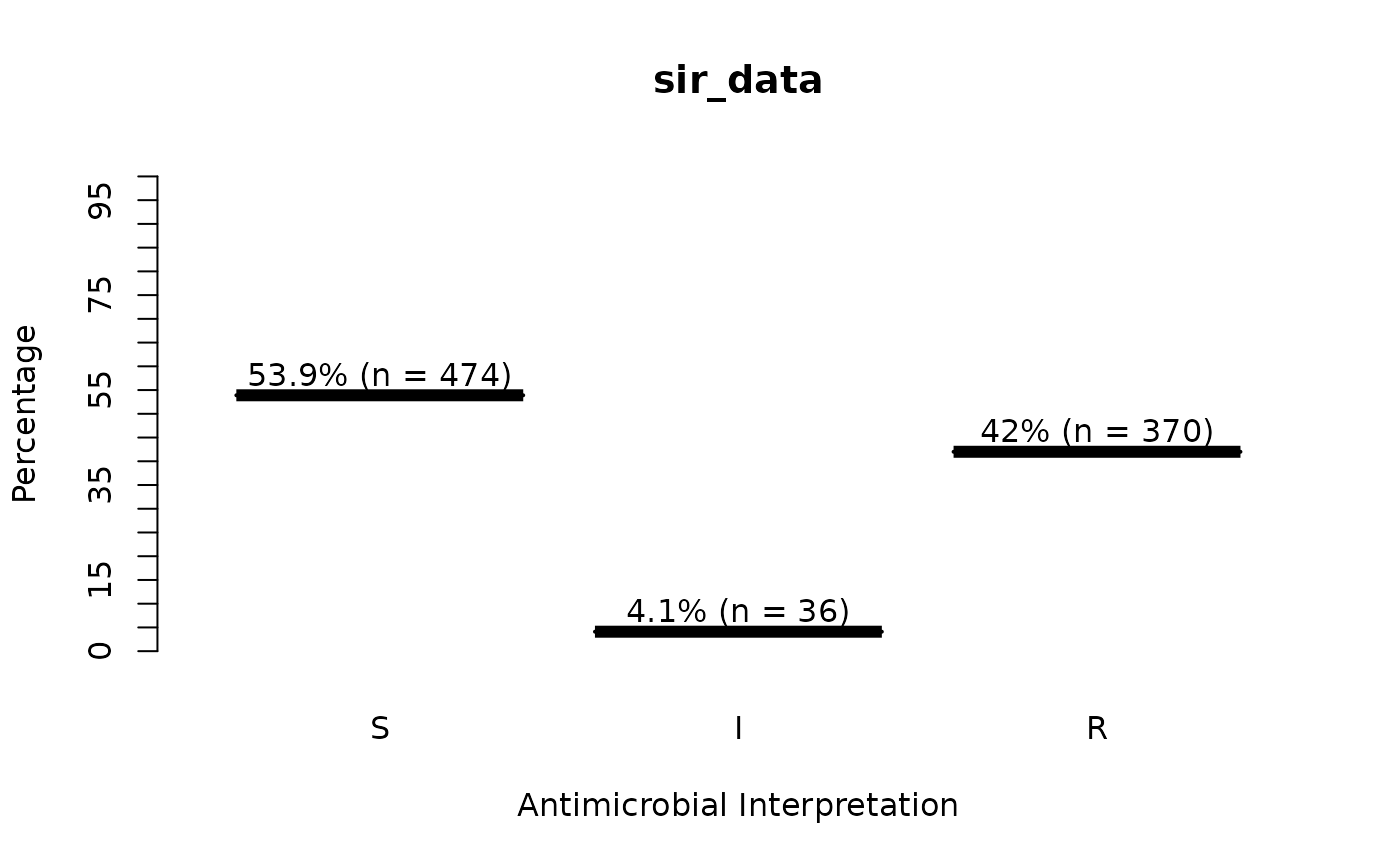
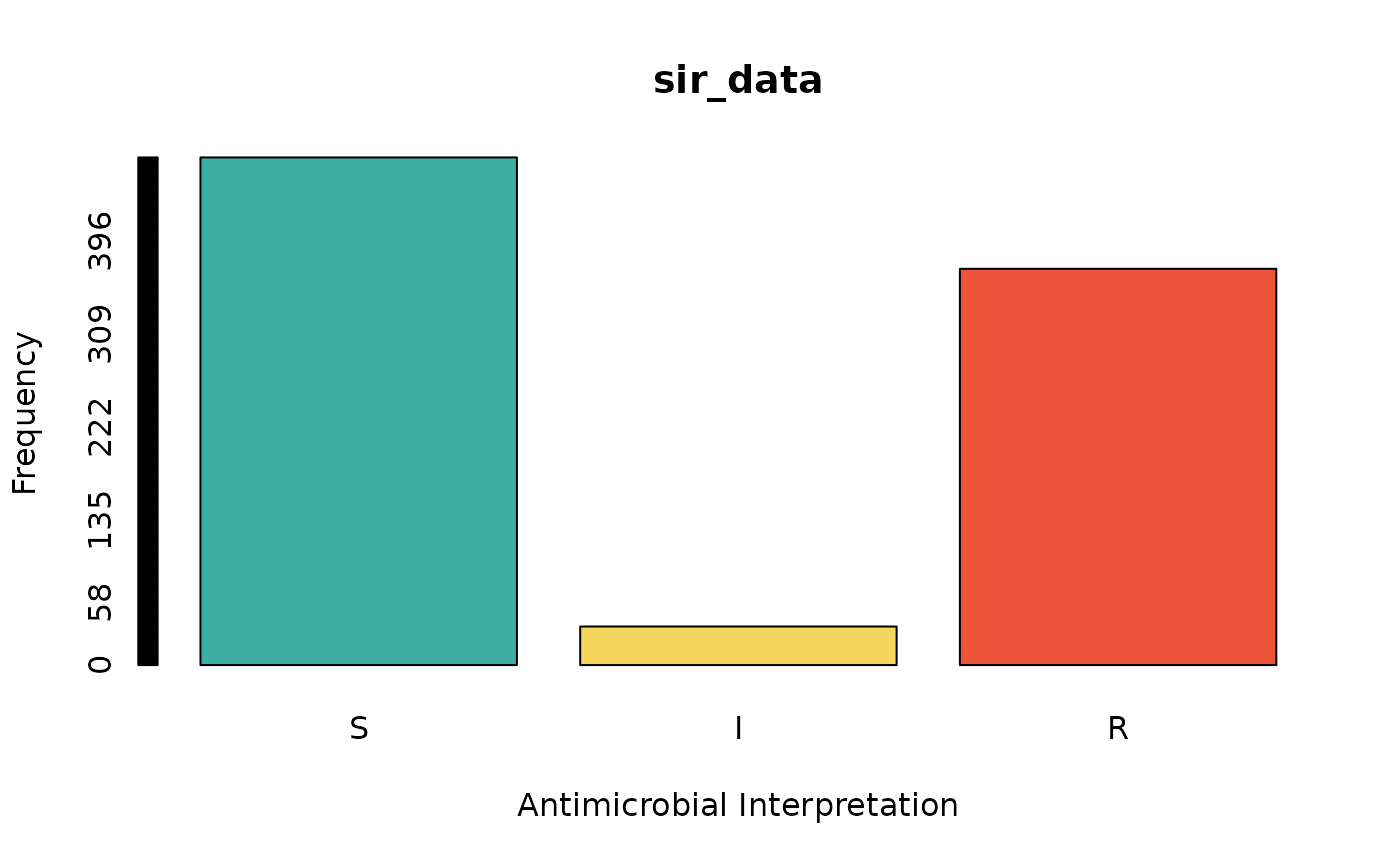
sir_data <- as.sir(c(rep("S", 474), rep("I", 36), rep("R", 370)))
is.sir(sir_data)
#> [1] TRUE
plot(sir_data) # for percentages
 barplot(sir_data) # for frequencies
barplot(sir_data) # for frequencies
 # as common in R, you can use as.integer() to return factor indices:
as.integer(as.sir(c("S", "SDD", "I", "R", "NI", NA)))
#> [1] 1 2 3 4 5 NA
# but for computational use, as.double() will return 1 for S, 2 for I/SDD, and 3 for R:
as.double(as.sir(c("S", "SDD", "I", "R", "NI", NA)))
#> [1] 1 2 2 3 NA NA
# the dplyr way
if (require("dplyr")) {
example_isolates %>%
mutate_at(vars(PEN:RIF), as.sir)
# same:
example_isolates %>%
as.sir(PEN:RIF)
# fastest way to transform all columns with already valid AMR results to class `sir`:
example_isolates %>%
mutate_if(is_sir_eligible, as.sir)
# since dplyr 1.0.0, this can also be the more impractical:
# example_isolates %>%
# mutate(across(where(is_sir_eligible), as.sir))
}
#> # A tibble: 2,000 × 46
#> date patient age gender ward mo PEN OXA FLC AMX
#> <date> <chr> <dbl> <chr> <chr> <mo> <sir> <sir> <sir> <sir>
#> 1 2002-01-02 A77334 65 F Clinical B_ESCHR_COLI R NA NA NA
#> 2 2002-01-03 A77334 65 F Clinical B_ESCHR_COLI R NA NA NA
#> 3 2002-01-07 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 4 2002-01-07 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 5 2002-01-13 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 6 2002-01-13 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 7 2002-01-14 462729 78 M Clinical B_STPHY_AURS R NA S R
#> 8 2002-01-14 462729 78 M Clinical B_STPHY_AURS R NA S R
#> 9 2002-01-16 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 10 2002-01-17 858515 79 F ICU B_STPHY_EPDR R NA S NA
#> # ℹ 1,990 more rows
#> # ℹ 36 more variables: AMC <sir>, AMP <sir>, TZP <sir>, CZO <sir>, FEP <sir>,
#> # CXM <sir>, FOX <sir>, CTX <sir>, CAZ <sir>, CRO <sir>, GEN <sir>,
#> # TOB <sir>, AMK <sir>, KAN <sir>, TMP <sir>, SXT <sir>, NIT <sir>,
#> # FOS <sir>, LNZ <sir>, CIP <sir>, MFX <sir>, VAN <sir>, TEC <sir>,
#> # TCY <sir>, TGC <sir>, DOX <sir>, ERY <sir>, CLI <sir>, AZM <sir>,
#> # IPM <sir>, MEM <sir>, MTR <sir>, CHL <sir>, COL <sir>, MUP <sir>, …
# }
# as common in R, you can use as.integer() to return factor indices:
as.integer(as.sir(c("S", "SDD", "I", "R", "NI", NA)))
#> [1] 1 2 3 4 5 NA
# but for computational use, as.double() will return 1 for S, 2 for I/SDD, and 3 for R:
as.double(as.sir(c("S", "SDD", "I", "R", "NI", NA)))
#> [1] 1 2 2 3 NA NA
# the dplyr way
if (require("dplyr")) {
example_isolates %>%
mutate_at(vars(PEN:RIF), as.sir)
# same:
example_isolates %>%
as.sir(PEN:RIF)
# fastest way to transform all columns with already valid AMR results to class `sir`:
example_isolates %>%
mutate_if(is_sir_eligible, as.sir)
# since dplyr 1.0.0, this can also be the more impractical:
# example_isolates %>%
# mutate(across(where(is_sir_eligible), as.sir))
}
#> # A tibble: 2,000 × 46
#> date patient age gender ward mo PEN OXA FLC AMX
#> <date> <chr> <dbl> <chr> <chr> <mo> <sir> <sir> <sir> <sir>
#> 1 2002-01-02 A77334 65 F Clinical B_ESCHR_COLI R NA NA NA
#> 2 2002-01-03 A77334 65 F Clinical B_ESCHR_COLI R NA NA NA
#> 3 2002-01-07 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 4 2002-01-07 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 5 2002-01-13 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 6 2002-01-13 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 7 2002-01-14 462729 78 M Clinical B_STPHY_AURS R NA S R
#> 8 2002-01-14 462729 78 M Clinical B_STPHY_AURS R NA S R
#> 9 2002-01-16 067927 45 F ICU B_STPHY_EPDR R NA R NA
#> 10 2002-01-17 858515 79 F ICU B_STPHY_EPDR R NA S NA
#> # ℹ 1,990 more rows
#> # ℹ 36 more variables: AMC <sir>, AMP <sir>, TZP <sir>, CZO <sir>, FEP <sir>,
#> # CXM <sir>, FOX <sir>, CTX <sir>, CAZ <sir>, CRO <sir>, GEN <sir>,
#> # TOB <sir>, AMK <sir>, KAN <sir>, TMP <sir>, SXT <sir>, NIT <sir>,
#> # FOS <sir>, LNZ <sir>, CIP <sir>, MFX <sir>, VAN <sir>, TEC <sir>,
#> # TCY <sir>, TGC <sir>, DOX <sir>, ERY <sir>, CLI <sir>, AZM <sir>,
#> # IPM <sir>, MEM <sir>, MTR <sir>, CHL <sir>, COL <sir>, MUP <sir>, …
# }