Benchmarks
Matthijs S. Berends
27 February 2019
benchmarks.RmdOne of the most important features of this package is the complete microbial taxonomic database, supplied by the Catalogue of Life. We created a function as.mo() that transforms any user input value to a valid microbial ID by using AI (Artificial Intelligence) combined with the taxonomic tree of Catalogue of Life.
Using the microbenchmark package, we can review the calculation performance of this function. Its function microbenchmark() runs different input expressions independently of each other and measures their time-to-result.
In the next test, we try to ‘coerce’ different input values for Staphylococcus aureus. The actual result is the same every time: it returns its MO code B_STPHY_AUR (B stands for Bacteria, the taxonomic kingdom).
But the calculation time differs a lot. Here, the AI effect can be reviewed best:
S.aureus <- microbenchmark(as.mo("sau"),
as.mo("stau"),
as.mo("staaur"),
as.mo("STAAUR"),
as.mo("S. aureus"),
as.mo("S. aureus"),
as.mo("Staphylococcus aureus"),
times = 10)
print(S.aureus, unit = "ms", signif = 3)
#> Unit: milliseconds
#> expr min lq mean median uq max neval
#> as.mo("sau") 16.70 16.80 21.50 16.8 17.50 56.6 10
#> as.mo("stau") 33.70 34.00 46.60 40.2 50.50 83.7 10
#> as.mo("staaur") 16.70 16.80 28.60 17.3 41.90 59.7 10
#> as.mo("STAAUR") 16.60 16.70 21.90 17.1 20.00 56.6 10
#> as.mo("S. aureus") 24.50 24.90 25.40 25.2 26.10 26.7 10
#> as.mo("S. aureus") 24.60 25.00 31.30 25.1 25.30 85.1 10
#> as.mo("Staphylococcus aureus") 7.37 7.42 8.17 7.5 8.27 11.8 10In the table above, all measurements are shown in milliseconds (thousands of seconds). A value of 5 milliseconds means it can determine 200 input values per second. It case of 100 milliseconds, this is only 10 input values per second. The second input is the only one that has to be looked up thoroughly. All the others are known codes (the first one is a WHONET code) or common laboratory codes, or common full organism names like the last one. Full organism names are always preferred.
To achieve this speed, the as.mo function also takes into account the prevalence of human pathogenic microorganisms. The downside is of course that less prevalent microorganisms will be determined less fast. See this example for the ID of Thermus islandicus (B_THERMS_ISL), a bug probably never found before in humans:
T.islandicus <- microbenchmark(as.mo("theisl"),
as.mo("THEISL"),
as.mo("T. islandicus"),
as.mo("T. islandicus"),
as.mo("Thermus islandicus"),
times = 10)
print(T.islandicus, unit = "ms", signif = 3)
#> Unit: milliseconds
#> expr min lq mean median uq max neval
#> as.mo("theisl") 289.0 291.0 314.0 312.0 336.0 343 10
#> as.mo("THEISL") 290.0 293.0 319.0 331.0 337.0 344 10
#> as.mo("T. islandicus") 73.5 73.8 90.6 75.1 114.0 118 10
#> as.mo("T. islandicus") 73.7 73.9 78.5 74.6 74.7 115 10
#> as.mo("Thermus islandicus") 66.5 67.3 80.3 69.5 107.0 108 10That takes 6.7 times as much time on average. A value of 100 milliseconds means it can only determine ~10 different input values per second. We can conclude that looking up arbitrary codes of less prevalent microorganisms is the worst way to go, in terms of calculation performance. Full names (like Thermus islandicus) are almost fast - these are the most probable input from most data sets.
In the figure below, we compare Escherichia coli (which is very common) with Prevotella brevis (which is moderately common) and with Thermus islandicus (which is very uncommon):
par(mar = c(5, 16, 4, 2)) # set more space for left margin text (16)
boxplot(microbenchmark(as.mo("Thermus islandicus"),
as.mo("Prevotella brevis"),
as.mo("Escherichia coli"),
as.mo("T. islandicus"),
as.mo("P. brevis"),
as.mo("E. coli"),
times = 50),
horizontal = TRUE, las = 1, unit = "s", log = FALSE,
xlab = "", ylab = "Time in seconds",
main = "Benchmarks per prevalence")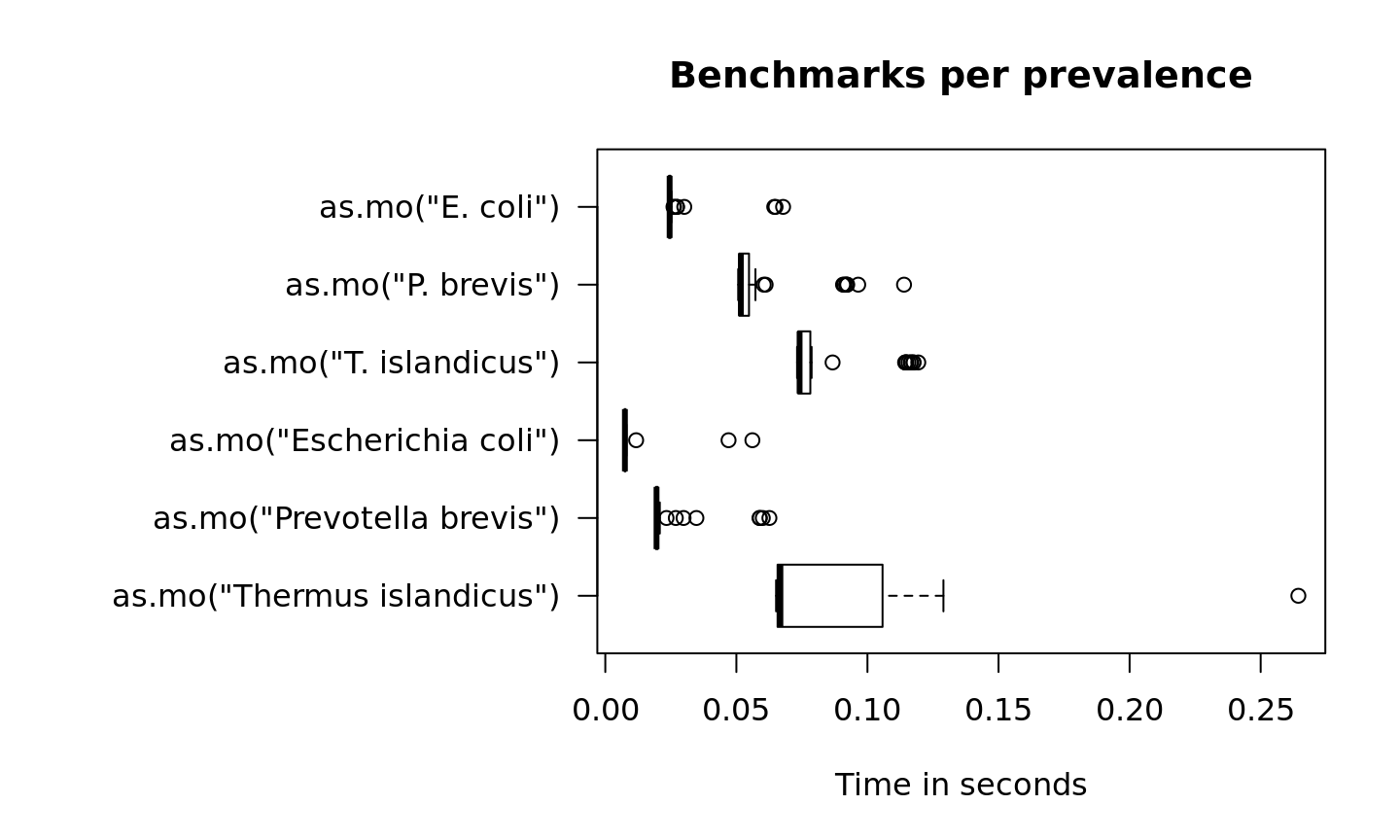
Uncommon microorganisms take a lot more time than common microorganisms. To relieve this pitfall and further improve performance, two important calculations take almost no time at all: repetitive results and already precalculated results.
Repetitive results
Repetitive results mean that unique values are present more than once. Unique values will only be calculated once by as.mo(). We will use mo_fullname() for this test - a helper function that returns the full microbial name (genus, species and possibly subspecies) which uses as.mo() internally.
library(dplyr)
# take all MO codes from the septic_patients data set
x <- septic_patients$mo %>%
# keep only the unique ones
unique() %>%
# pick 50 of them at random
sample(50) %>%
# paste that 10,000 times
rep(10000) %>%
# scramble it
sample()
# got indeed 50 times 10,000 = half a million?
length(x)
#> [1] 500000
# and how many unique values do we have?
n_distinct(x)
#> [1] 50
# now let's see:
run_it <- microbenchmark(mo_fullname(x),
times = 10)
print(run_it, unit = "ms", signif = 3)
#> Unit: milliseconds
#> expr min lq mean median uq max neval
#> mo_fullname(x) 732 768 800 794 799 949 10So transforming 500,000 values (!!) of 50 unique values only takes 0.79 seconds (793 ms). You only lose time on your unique input values.
Precalculated results
What about precalculated results? If the input is an already precalculated result of a helper function like mo_fullname(), it almost doesn’t take any time at all (see ‘C’ below):
run_it <- microbenchmark(A = mo_fullname("B_STPHY_AUR"),
B = mo_fullname("S. aureus"),
C = mo_fullname("Staphylococcus aureus"),
times = 10)
print(run_it, unit = "ms", signif = 3)
#> Unit: milliseconds
#> expr min lq mean median uq max neval
#> A 10.900 11.300 11.900 11.500 11.700 15.200 10
#> B 22.000 22.600 26.900 22.600 22.800 62.400 10
#> C 0.311 0.502 0.531 0.533 0.577 0.765 10So going from mo_fullname("Staphylococcus aureus") to "Staphylococcus aureus" takes 0.0005 seconds - it doesn’t even start calculating if the result would be the same as the expected resulting value. That goes for all helper functions:
run_it <- microbenchmark(A = mo_species("aureus"),
B = mo_genus("Staphylococcus"),
C = mo_fullname("Staphylococcus aureus"),
D = mo_family("Staphylococcaceae"),
E = mo_order("Bacillales"),
F = mo_class("Bacilli"),
G = mo_phylum("Firmicutes"),
H = mo_kingdom("Bacteria"),
times = 10)
print(run_it, unit = "ms", signif = 3)
#> Unit: milliseconds
#> expr min lq mean median uq max neval
#> A 0.274 0.333 0.417 0.412 0.481 0.611 10
#> B 0.337 0.353 0.403 0.411 0.438 0.461 10
#> C 0.336 0.362 0.530 0.528 0.682 0.755 10
#> D 0.263 0.316 0.340 0.331 0.360 0.474 10
#> E 0.252 0.326 0.326 0.329 0.345 0.389 10
#> F 0.250 0.300 0.319 0.327 0.351 0.351 10
#> G 0.240 0.246 0.288 0.277 0.325 0.351 10
#> H 0.242 0.257 0.304 0.294 0.332 0.395 10Of course, when running mo_phylum("Firmicutes") the function has zero knowledge about the actual microorganism, namely S. aureus. But since the result would be "Firmicutes" too, there is no point in calculating the result. And because this package ‘knows’ all phyla of all known bacteria (according to the Catalogue of Life), it can just return the initial value immediately.
Results in other languages
When the system language is non-English and supported by this AMR package, some functions will have a translated result. This almost does’t take extra time:
mo_fullname("CoNS", language = "en") # or just mo_fullname("CoNS") on an English system
#> [1] "Coagulase Negative Staphylococcus (CoNS)"
mo_fullname("CoNS", language = "es") # or just mo_fullname("CoNS") on a Spanish system
#> [1] "Staphylococcus coagulasa negativo (CoNS)"
mo_fullname("CoNS", language = "nl") # or just mo_fullname("CoNS") on a Dutch system
#> [1] "Coagulase-negatieve Staphylococcus (CNS)"
run_it <- microbenchmark(en = mo_fullname("CoNS", language = "en"),
de = mo_fullname("CoNS", language = "de"),
nl = mo_fullname("CoNS", language = "nl"),
es = mo_fullname("CoNS", language = "es"),
it = mo_fullname("CoNS", language = "it"),
fr = mo_fullname("CoNS", language = "fr"),
pt = mo_fullname("CoNS", language = "pt"),
times = 10)
print(run_it, unit = "ms", signif = 4)
#> Unit: milliseconds
#> expr min lq mean median uq max neval
#> en 14.68 15.11 15.28 15.27 15.34 16.21 10
#> de 23.53 23.65 24.04 23.82 24.63 24.88 10
#> nl 23.36 23.53 27.76 23.81 23.87 63.45 10
#> es 23.59 23.83 23.94 23.96 24.07 24.22 10
#> it 23.30 23.76 27.92 23.83 24.18 64.01 10
#> fr 23.47 23.78 31.80 23.84 24.90 63.25 10
#> pt 23.28 23.69 27.97 23.85 24.42 64.14 10Currently supported are German, Dutch, Spanish, Italian, French and Portuguese.