How to conduct principal component analysis (PCA) for AMR
Matthijs S. Berends
15 April 2020
Source:vignettes/PCA.Rmd
PCA.RmdNOTE: This page will be updated soon, as the pca() function is currently being developed.
Transforming
For PCA, we need to transform our AMR data first. This is what the example_isolates data set in this package looks like:
library(AMR) library(dplyr) glimpse(example_isolates) # Rows: 2,000 # Columns: 49 # $ date <date> 2002-01-02, 2002-01-03, 2002-01-07, 2002-01-07, 2002… # $ hospital_id <fct> D, D, B, B, B, B, D, D, B, B, D, D, D, D, D, B, B, B,… # $ ward_icu <lgl> FALSE, FALSE, TRUE, TRUE, TRUE, TRUE, FALSE, FALSE, T… # $ ward_clinical <lgl> TRUE, TRUE, FALSE, FALSE, FALSE, FALSE, TRUE, TRUE, F… # $ ward_outpatient <lgl> FALSE, FALSE, FALSE, FALSE, FALSE, FALSE, FALSE, FALS… # $ age <dbl> 65, 65, 45, 45, 45, 45, 78, 78, 45, 79, 67, 67, 71, 7… # $ gender <chr> "F", "F", "F", "F", "F", "F", "M", "M", "F", "F", "M"… # $ patient_id <chr> "A77334", "A77334", "067927", "067927", "067927", "06… # $ mo <mo> B_ESCHR_COLI, B_ESCHR_COLI, B_STPHY_EPDR, B_STPHY_EPDR… # $ PEN <rsi> R, R, R, R, R, R, R, R, R, R, R, R, R, R, R, R, R, R,… # $ OXA <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ FLC <rsi> NA, NA, R, R, R, R, S, S, R, S, S, S, NA, NA, NA, NA,… # $ AMX <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ AMC <rsi> I, I, NA, NA, NA, NA, S, S, NA, NA, S, S, I, I, R, I,… # $ AMP <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ TZP <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ CZO <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ FEP <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ CXM <rsi> I, I, R, R, R, R, S, S, R, S, S, S, S, S, NA, S, S, R… # $ FOX <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ CTX <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, S, S,… # $ CAZ <rsi> NA, NA, R, R, R, R, R, R, R, R, R, R, NA, NA, NA, S, … # $ CRO <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, S, S,… # $ GEN <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ TOB <rsi> NA, NA, NA, NA, NA, NA, S, S, NA, NA, NA, NA, S, S, N… # $ AMK <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ KAN <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ TMP <rsi> R, R, S, S, R, R, R, R, S, S, NA, NA, S, S, S, S, S, … # $ SXT <rsi> R, R, S, S, NA, NA, NA, NA, S, S, NA, NA, S, S, S, S,… # $ NIT <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ FOS <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ LNZ <rsi> R, R, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, R, R, R… # $ CIP <rsi> NA, NA, NA, NA, NA, NA, NA, NA, S, S, NA, NA, NA, NA,… # $ MFX <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ VAN <rsi> R, R, S, S, S, S, S, S, S, S, NA, NA, R, R, R, R, R, … # $ TEC <rsi> R, R, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, R, R, R… # $ TCY <rsi> R, R, S, S, S, S, S, S, S, I, S, S, NA, NA, I, R, R, … # $ TGC <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ DOX <rsi> NA, NA, S, S, S, S, S, S, S, NA, S, S, NA, NA, NA, R,… # $ ERY <rsi> R, R, R, R, R, R, S, S, R, S, S, S, R, R, R, R, R, R,… # $ CLI <rsi> NA, NA, NA, NA, NA, R, NA, NA, NA, NA, NA, NA, NA, NA… # $ AZM <rsi> R, R, R, R, R, R, S, S, R, S, S, S, R, R, R, R, R, R,… # $ IPM <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, S, S,… # $ MEM <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ MTR <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ CHL <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ COL <rsi> NA, NA, R, R, R, R, R, R, R, R, R, R, NA, NA, NA, R, … # $ MUP <rsi> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N… # $ RIF <rsi> R, R, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, R, R, R…
Now to transform this to a data set with only resistance percentages per taxonomic order and genus:
resistance_data <- example_isolates %>% group_by(order = mo_order(mo), # group on anything, like order genus = mo_genus(mo)) %>% # and genus as we do here summarise_if(is.rsi, resistance) %>% # then get resistance of all drugs select(order, genus, AMC, CXM, CTX, CAZ, GEN, TOB, TMP, SXT) # and select only relevant columns head(resistance_data) # # A tibble: 6 x 10 # # Groups: order [2] # order genus AMC CXM CTX CAZ GEN TOB TMP SXT # <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> # 1 (unknown orde… Micrococcoides NA NA NA NA NA NA NA NA # 2 Actinomycetal… Actinomyces NA NA NA NA NA NA NA NA # 3 Actinomycetal… Corynebacterium NA NA NA NA NA NA NA NA # 4 Actinomycetal… Dermabacter NA NA NA NA NA NA NA NA # 5 Actinomycetal… Micrococcus NA NA NA NA NA NA NA NA # 6 Actinomycetal… Propionibacter… NA NA NA NA NA NA NA NA
Perform principal component analysis
The new pca() function will automatically filter on rows that contain numeric values in all selected variables, so we now only need to do:
pca_result <- pca(resistance_data) # NOTE: Columns selected for PCA: AMC/CXM/CTX/CAZ/GEN/TOB/TMP/SXT. # Total observations available: 7.
The result can be reviewed with the good old summary() function:
summary(pca_result) # Importance of components: # PC1 PC2 PC3 PC4 PC5 PC6 PC7 # Standard deviation 2.1580 1.6783 0.61282 0.33017 0.20150 0.03190 2.123e-16 # Proportion of Variance 0.5821 0.3521 0.04694 0.01363 0.00508 0.00013 0.000e+00 # Cumulative Proportion 0.5821 0.9342 0.98117 0.99480 0.99987 1.00000 1.000e+00
Good news. The first two components explain a total of 93.4% of the variance (see the PC1 and PC2 values of the Proportion of Variance. We can create a so-called biplot with the base R biplot() function, to see which antimicrobial resistance per drug explain the difference per microorganism.
Plotting the results
biplot(pca_result)
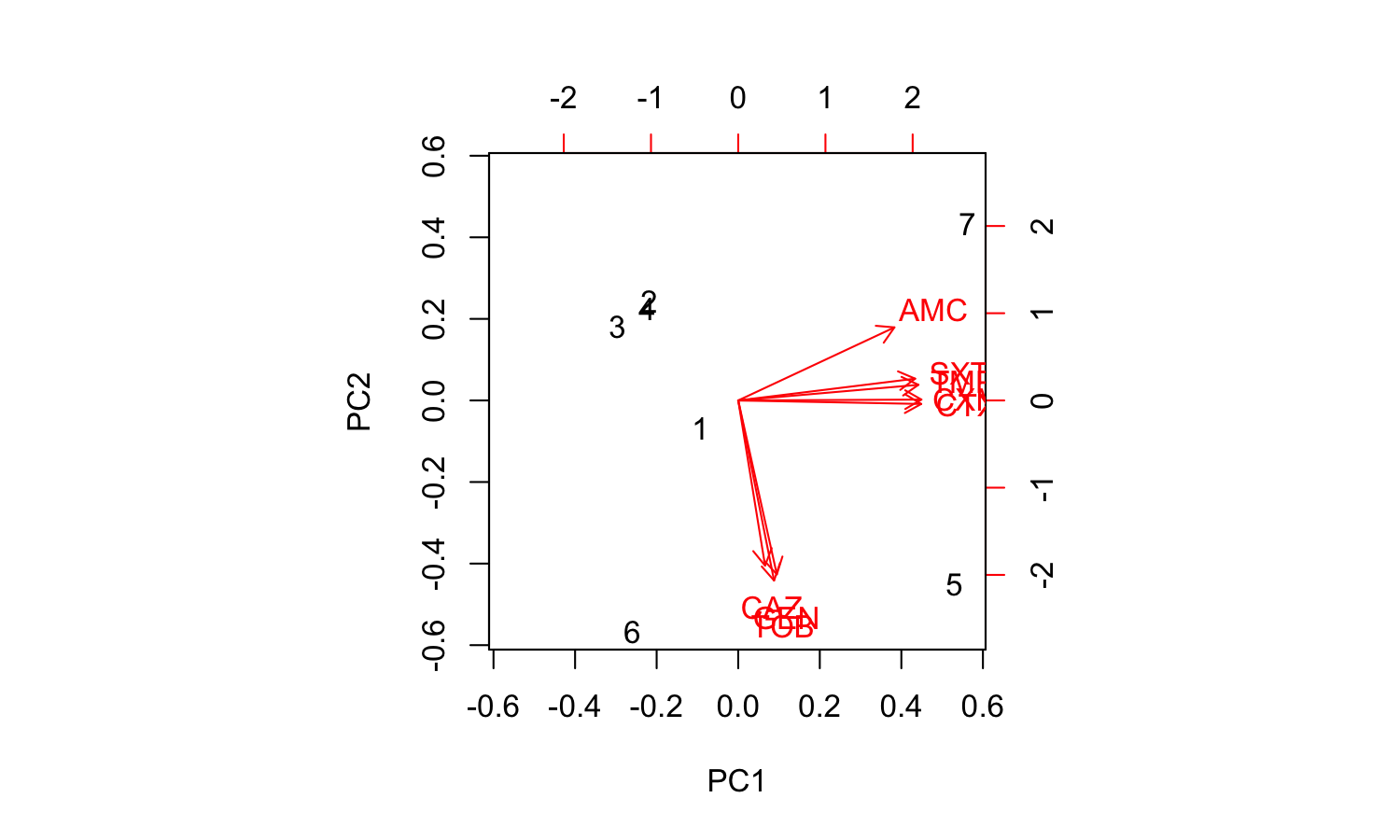
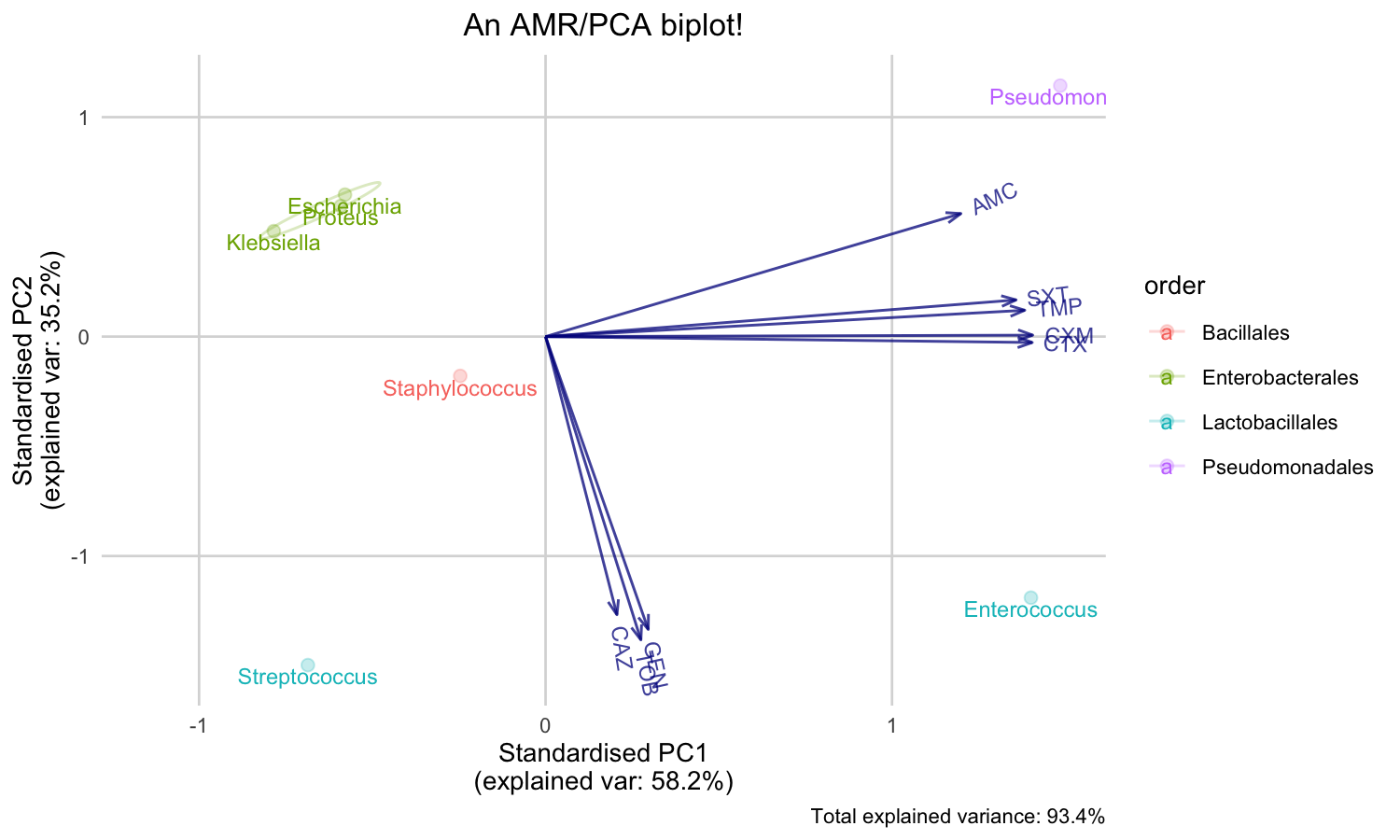
But we can’t see the explanation of the points. Perhaps this works better with our new ggplot_pca() function, that automatically adds the right labels and even groups:
ggplot_pca(pca_result)
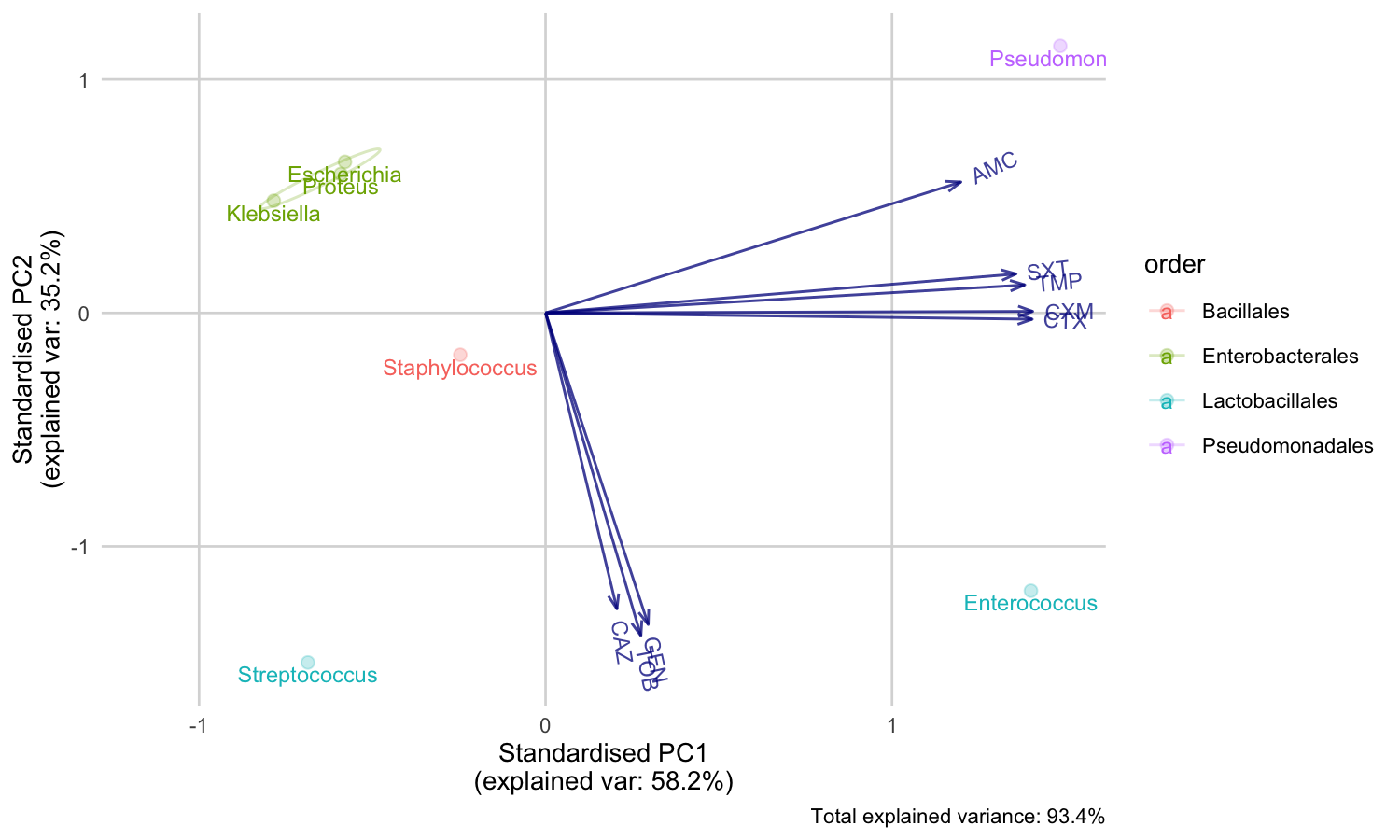
You can also print an ellipse per group, and edit the appearance:
ggplot_pca(pca_result, ellipse = TRUE) + ggplot2::labs(title = "An AMR/PCA biplot!")